I like this post and I thought that a way to "play along productively" might be to take a crack at Alzheimer's on my own and narrate the process a bit?
Then maybe see if this piece of advice can be applied?
What you won't feel is a sense of building understanding. Learning to notice a lack of understanding is one of the most important skills, and it is sadly not an easy thing to explain.
I tried several approaches to figuring out what might be going on.
First, I looked for "part of the brain" or "type of cell" signatures that could distinguish between "a broken thing causes the symptoms" and "nearly any thing broken causes the larger working system to fail". Seeking "parts and such" popular pres summaries suggested maybe the entorhinal cortex is the first thing to look like it is damaged. Then imaging studies with citable research seems to agree... but nothing "pops out" to me, so I moved on. (Maybe something will loop back somehow?)
Second, I thought that maybe if risk factors could say anything about pre-disposing factors or mechanistic harbingers of onset, that might help generate a list of candidate hypotheses. It turns out someone DID perform a cohort study of healthy people to watch some (but not all) get the symptoms. Figure 3 was helpful in showing that lots of little things can all be used to improve predictions of who will get it, but nothing beyond the basic model is really decisive:

Mentally "double-clicking" on the APOE connection (because that's part of the mostly-adequate basic model) some researchers made human stem cells from people with different alleles and then turned them into neurons that should or should not hypothetically be different on this axis and then played with them. Something something phosolipid metabolism? Adding the alzheimer's APOE allele to yeast made the yeast weirdly fatty too and then giving the predisoposed yeast more of the nutrient choline sort of made then go back to "looking healthy"?
This was my first "feeling of recognition" where a test of "a building sense of understanding" could happen, because choline has come up for me in the past!
Acetylcholine is a neurotransmitter oft modulated by the 'racetam family of nootropics, which can cause brain fog after or during the nootropic use, and this is anecdotally ameliorated by taking choline before or during. (Sort of like with tryptophan and ecstasy maybe, but plausibly a similar sort of epistemic cesspool.)
The double coincidence of "maybe fixing bad brain stuff by taking choline" is suggestive to me?
My goto source for choline is simply eating eggs, whose yolks are rich in choline. Googling [egg yolk's alzheimers] gave a directionally positive hit, (which is bayesian evidence for not being totally confused) but looking at it mechanistically, it seems like maybe the egg yolks (roughly) were helping with frontal cortex health, and then the frontal cortex was helping people cope with dementia, but dementia onset itself was not prevented by eating food with clever nutrients?
Time to shake the bag and draw a new marble!
...
My final (more precise) shot in the dark (this time on Bing because Google might be getting shitty these days) is based on the stuff above: [phospholipid metabolism entorhinal cortex imaging onset]. The first hit was a paper, and the second was a suggestion to try an image search, I did both in parallel and got wildly divergent suggestions about what was salient.
The image search was interesting because most of the hits had associated text that didn't even MENTION Alzheimer's. The fifth image did mention AD, which suggested that I hadn't gone totally off the conceptual reservation yet, but most hits were to things like: "hypoparathyroidism mimicking ankylosing spondylitis" and "myoclonus" and "juevenile onset hypokinetic-rigid Huntingtons" and "Adult-onset leukoencephalopathy with calcifications" and so on. (NOTE: the bolded "calcification" will make sense further along.)
So like one way to imagine this being "at all helpful" is to imagine that maybe any super high quality model of AD would just be a fully general model of brain health?
Using a highly general, high quality model would enable knowing someone had AD but not any of these other things... and then turning the model's crank to imply things that were still going RIGHT in AD brains (because the AD brain doesn't have the other diseases too, at the same time?) and those things going RIGHT would be things that it'd be safe to remove from a model of AD itself?
A really strong model here would thus enable one to notice "dogs not barking" which give positive evidence about the state of the brain of an AD patient who does not also have those other symptoms and thus must have parts of their brain that are relatively healthy?
If these parts are healthy when AD symptoms present clinically, that at least narrows the search space! However, building a model able to mechanically perform such inferences-from-absense-of-unusual-evidence seems like it would take a LOT of elbow grease. No thanks for now... shake the bag, draw another marble?
Yet also... the #1 Bing search result was interesting and daunting... and maybe led to paydirt?
I got a "connection to something I've heard of before" in the form of links to DHA and EPA which are nutritionally interesting (it is fish fat stuff, basically (and a guy named Dr. Sears tried to save the world with a diet and supplement system based on this aspect of metabolism)). Prolly not specific enough.
But Bing's #1 hit did suggest that the levels of this stuff decline with "healthy" aging but also that they concentrate more and more into the membranes of the mitochondria, presumably leaving their neurons's outer membranes relatively denuded of these substances (in order to get the total to decline)?
So what might be happening here is the "most valuable things" (the mitochondria) are being preserved over time as people engage in "healthy aging" (which is sort of an oxymoron if you think of it... its just coping as well as most people with intrinsic sadness... which is not the same thing as happy thriving). This is causing my spider sense to tingle with a maybe not super dumb concordance?
Mitochondria are super interesting in general, and I've had past strong reasons to suppose that aging causes them to decline in a way that is mostly orthogonal to EPA or DHA (at least orthogonal so far as I've ever heard)... and then in general "mitochondria repair or replacement" is, in my opinion, going to be an essential subtherapy in any actually working SENS treatment regime.
So on a hunch, I went looking for a NEW search term and this is where I hit paydirt maybe?
The search was [mitochondrial haplogroup alzheimer’s differences] and the hit was Wang & Brinton's fantastic review paper that just spells out in detail many concrete details I was sort of hoping to find which I will hereby call "The Swerdlow & Khan Mitochondrial Cascade Hypothesis For Late Onset Alzheimer’s Disease" or for short the MC->LOAD Hypothesis.
One of the things I love about this is that it resonates with the giant model that could notice "quiet dogs"!
First, there's a whole collection of things that NEED energy that can fail. Maybe whichever "energy-hungry thing downstream of mitochondrial health" is least efficient in a specific person is the one that will start to "tell" first, maybe also leading doctors to observe different things and create different names for different apparent diseases?
This is where an earlier mention of "calcification" seemed like a hint that the whole thing might be tractable-in-general with sufficient elbow grease?
Wang & Brinton summarize some of the nodes you might want in a very big Pearlian graph about neuron mechanism thusly:
mitochondrial genetic variations and mutations lead to deficient electron transport chain function, resulting in less ATP production, increased free radical production, disrupted calcium homeostasis, beta amyloid production and plaque deposition, and tau phosphorylation and tangle formation. These results in turn lead to further damage of mitochondrial DNA, proteins, and lipids, and the opening of mitochondrial permeability transition pore, which ultimately leads to cell death and neurodegeneration
Their Figure 1 even has a "nodes with text and arrows between the concepts" presentation!
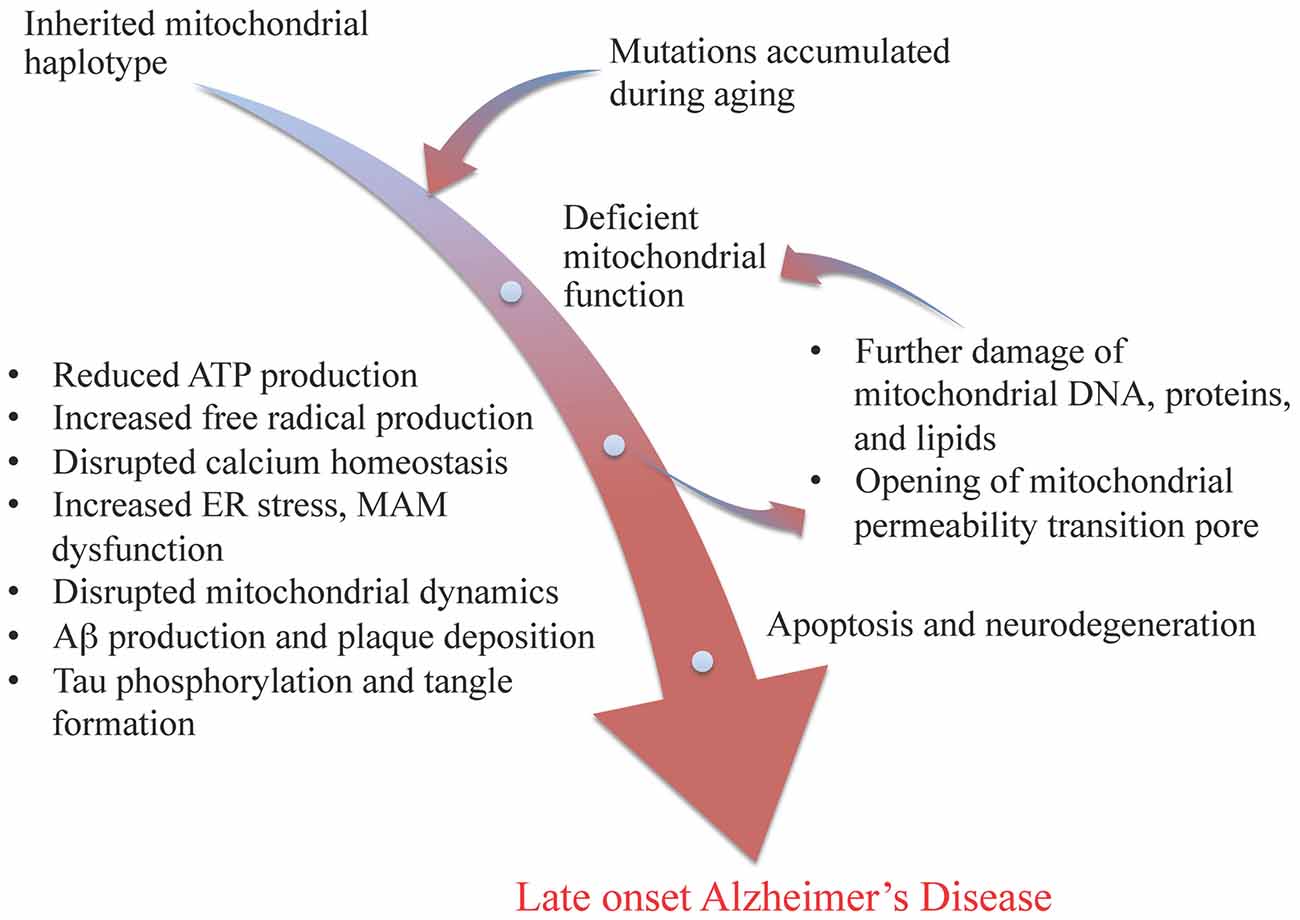
The paper this is from has numerous indicators of high quality (at least in my opinion).
It explains lots of systems in a nuts and bolts way. It baldly asserts things that I would expect others to hedge on, but which accord with my common sense. It displays clear hallmarks of an approach where "the dominoes just fall over somehow, and our job is to figure out the details, and make and test predictions about what else they might knock over... while they fall however they fall".
Something that jumped out to me as potentially helpful here was a (sadly small (N=36)) study finding that proxies of mitochondrial efficiency were lower in people whose mother's had had Late Onset Alzheimer's Disease. A new kind of heritability would be mechanistically important!
Maybe it won't replicate? The dots aren't very numerous, and the separation of people with a Maternal History (MH) of LOAD isn't perfect:

So yeah. Maybe I made my brain dumber by exposing it to low quality stuff, but this model feels like it homes in on more foundational mechanisms: like mitochondria!
Another thing I think I've homed in on is Swerdlow who (if I read the author list right) was the PI for the Mosconi study and an early articulator of this hypothesis.
Looping ALL THE WAY BACK... the exercise was focused on this skill:
What you won't feel is a sense of building understanding. Learning to notice a lack of understanding is one of the most important skills, and it is sadly not an easy thing to explain.
I feel like maybe this is a place where I can summon up various sort "socio-cultural models of evidentiary integrity" where I could try to:
(1) brag about doing this well (like: Swerdlow's hypothesis sorta explains why amyloid reduction isn't addressing the core problem and might not help with clinical outcomes (like if the cell's energy budget is no longer penciling out, the cell is going to die of something and amyloid buildup might be a ONE thing to tolerate-for-a-while as budget shortfalls cause maintenance skips)) or
(2) engage in paranoid speculation about Swerdlow's lab maybe p-hacking with small n studies and a citation clique (but honestly who can afford large N or wants to avoid doing research with friends) such that maybe most science is false and none of it will replicate, or
(3) just try to be humble and push my hands very wide and say "my error bars are still very wide", and justify my error bars with laziness, because the ideal causal graph would take more elbow grease than I have time to build?
A hilariously simple test (to cut through some of the bullshit) might be to just like... inject a bunch of high quality living mitochondria into someone's veins as an "organelle transplant" and see what happens? It kinda seems to work in rats for other things? If it fails, that wouldn't be dispositive (maybe the needle missed the vein, or the immune system fought back, or maybe other problems... etc) but if it works it would seem to clinch both the theory and that specific method of fixing what the theory says is wrong.
This is an excellent comment, and I'm very glad to see my thinking inspiring others!
My own findings on the issue are as such:
I am confident that mitochondrial dysfunction is upstream of AD.
This one gene called PGC 1 alpha is probably involved or something.
I do not know what (if anything) is upstream of that. It could be immune system health but the immune system is so complex that my understanding of it is generally poor.
Mitochondria which are defective are replaced in cells through a process called mitophagy. Stimulating the creation of mitochondria (mitogenesis) probably increases mitophagy too as cells can regulate themselves pretty well.
Drugs which stimulate mitophagy and mitogenesis are probably more productive avenues for AD research than lots of things, an example of each is metformin and EET-A respectively. EET-A has an effect on amyloid plaques in mice but that isn't that useful (however I think that information is worth more when a mechanistic story can be told).
I suspect that AD is an "attractive state" that brains just fall into for lots of reasons, this explains the endless feedback loops confusing researchers, and also it being much more common than lots of other brain diseases.
AD is an interesting microcosm of not just brain health, but also of ageing in general. An effective anti-ageing therapy would almost certainly eliminate AD or at least stop progression. An effective AD therapy is worth investigating as a general anti-ageing therapy (although if it's that effective on ageing we'll probably notice).
Alzheimer's Disease (AD) is truly, unduly cruel, and truly, unduly common. A huge amount of effort goes into curing it, which I think is a true credit to our civilization. This is in the form of both money, and the efforts of many of the brightest researchers.
But it hasn't worked.
Since AD is characterised by amyloid plaques, the "amyloid hypothesis" that these were the causative agent has been popular for a while. Mutations to genes which encode the amyloid beta protein can cause AD. Putting lots of amyloid into the brain causes brain damage in mice. So for many years, drugs were screened by testing them in mutant mice which were predisposed to AD. If the plaques disappeared, they were considered good candidates.
So why didn't it work?
Lots of things can affect amyloid plaques as it turns out, right up to the latest FDA approved drug, which is just antibodies which target amyloid protein. While this does reduce amyloid, it has no effect on cognitive decline.
Goodhart's law has reared its head: amyloid plaque buildup is a metric for AD progression, but selecting for drugs which reduce it causes the relationship between AD and plaques to fall apart.
Equally, amyloid plaques are very easy to measure in mouse (and human) brains. It can be done by MRI scan, or by dissection. Memory loss and mood changes are harder to measure, and even harder in mice. The methods for measuring amyloid plaques also feel better in many ways. There's less variation in potential methods, they can be compared across species, they're qualitative, and they're also more in line with what the average biologist/chemist will be used to.
Understanding these, we can see how looking for drugs which decrease amyloid plaques in mice just really feels like productive research. We can also understand, now, why it wasn't.
Avoiding Wasted Effort
Pointing out biases is fairly useless. Pointing out specific examples is better. But the best way to help others is to point out how it feels from the inside to be making these mistakes.
So what does it feel like to be on the inside of these biases? Unfortunately as someone who has not been intimately involved in AD research I can't say exactly. But as someone involved with research in general I can make a guess:
Beyond this I do not know. Perhaps it is a nameless virtue. But it might be useful to try to identify more cases. I hereby precommit to posting a follow-up with at least five examples of this within the next seven days.