Whether an individual develops schizophrenia, obesity, diabetes, or autism depends more on their genes (and epi-genome) than it does on any other factor.
Would appreciate a source for this assertion.
I chatted with Michael and Ammon. This made me somewhat more hopeful about this effort, because their plan wasn't on the less-sensible end of what I uncertainly imagined from the post (e.g. they're not going to just train a big very-nonlinear map from genomes to phenotypes, which by default would make the data problem worse not better).
I have lots of (somewhat layman) question marks about the plan, but it seems exciting/worth trying. I hope that if there are some skilled/smart/motivated/curious ML people seeing this, who want to work on something really cool and/or that could massively help the world, you'll consider reaching out to Tabula.
An example of the sort of thing they're planning on trying:
1: Train an autoregressive model on many genomes as base-pair sequences, both human and non-human. (Maybe upweight more-conserved regions, on the theory that they're conserved because under more pressure to be functional, hence more important for phenotypes.)
1.5: Hope that this training run learns latent representations that make interesting/important features more explicit.
2: Train a linear or linear-ish predictor from the latent activations to some phenotype (disease, personality, IQ, etc.).
IDK if I expect this to work well, but it seems like it might. Some question marks:
- Do we have enough data? If we're trying to understand rare variants, we want whole genome sequences. My quick convenience sample turned up about 2 million publicly announced whole genomes. Maybe there's more like 5 million, and lots of biobanks have been ramping up recently. But still, pretending you have access to all this data, this means you see any given region a couple million times. Not sure what to think of that; I guess it depends what/how we're trying to learn.
- If we focus on conserved regions, we probably successfully pull attention away from regions that really don't matter. But we might also pull attention away from regions that sorta matter, but to which phenotypes of interest aren't terribly sensitive to. It stands to reason that such regions or variants wouldn't be the most conserved regions or variants. I don't think this defeats the logic, but it suggests we could maybe do better. Example of other approaches: upvote regions that are recognized as having the format of genes or regulatory regions; upvote regions around SNPs that have shown up in GWASes.
- For the exact setup described above, with autoregression on raw genomes, do we really learn much about variants? I guess what we ought to learn something about is approximate haplotypes, i.e. linkage disequilibrium structure. The predictor should be like "ok, at the O(10kb) moment, I'm in such-and-such gene regulatory module, and I'm looking at haplotype number 12 for this region, so by default I should expect the following variants to be from that haplotype, unless I see a couple variants that are rarely with H12 but all come from H5 or something". I don't see how this would help identify causal SNPs out of haplotypes? But it could very well make the linear regression problem significantly easier? Not sure.
- But also I wouldn't expect this exact setup to tell us much interesting stuff about anything long-range, e.g. protein-protein interactions. Well, on the other hand, possibly there'd be some shared representation between "either DNA sequence X; or else a gene that codes for a protein that has a sequence of zinc fingers that match up with X"? IDK what to expect. This could maybe be enhanced with models of protein interactions, transcription factor binding affinities, activity of regulatory regions, etc. etc.
More generally, I'm excited about someone making a concerted and sane effort to try putting biological priors to use for genomic predictions. As a random example (which may not make much sense, but to give some more flavor): Maybe one could look at AlphaFold's predictions of protein conformation with different rare genetic variants that we've marked as deleterious for some trait. If the predictions are fairly similar for the different variants, we don't conclude much--maybe this rare variant has some other benefit. But if the rare variant makes AlphaFold predict "no stable conformation", then we take this as some evidence that the rare variant is purely deleterious, and therefore especially safe to alter to the common variant.
I'm not as concerned about your points because there are a number of projects already doing something similar and (if you believe them) succeeding at it. Here's a paper comparing some of them: https://www.biorxiv.org/content/10.1101/2025.02.11.637758v2.full
Congrats on the new company! I think this is potentially quite an important effort, so I hope anyone browing this forum who has a way to get access to biobank data from various sources will reach out.
One of my greatest hopes for these new models is that they will provide a way for us to predict the effects of novel genetic variants on traits like intelligence or disease risk.
When I wrote my post on adult intelligence enhancement at the end of 2023, one of the biggest issues was just how many edits we would need to make to achieve significant change in intelligence.
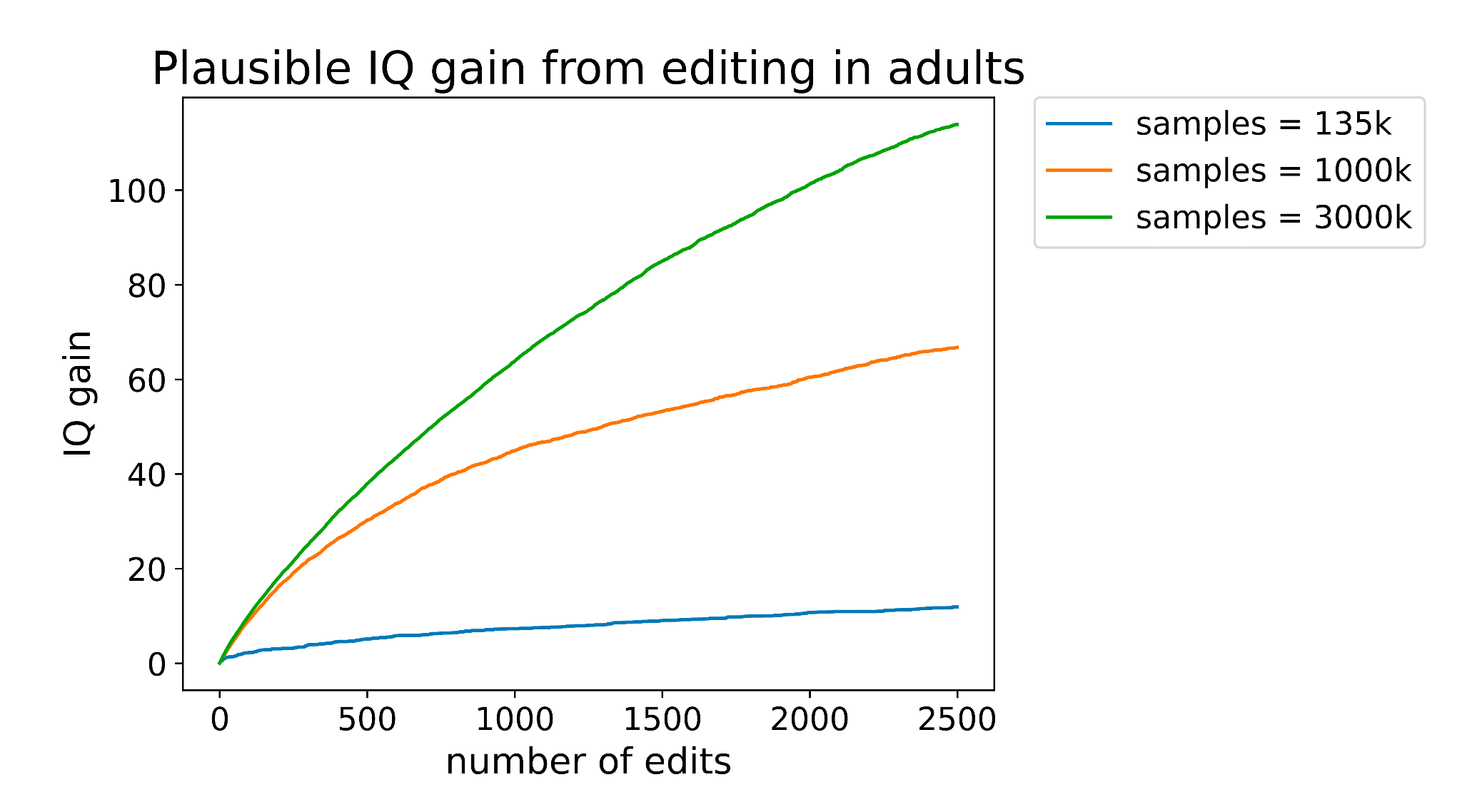
Neither delivery nor editing tech is good enough yet to make anywhere close to the number of edits needed for adult cognitive enhancement. But it's plausible that there exist a set of gene edits such that just 10 or 20 changes would have a significant impact.
One of my greatest hopes is that these foundation models will generalize well enough out-of-distribution that we can make reasonably accurate predictions about the effect of NEW genetic variants on traits like intelligence of health. If we can, (and ESPECIALLY if we can also predict tolerance to these edits), it could 10x the impact of gene editing.
This may end up being one of the most important technologies for adult cognitive enhancement. So I hope anyone who might be able to help with data access reaches out to you!
Congrats on the launch -- the pitch is certainly super exciting.
Approximately all AI-bio tools are worryingly dual use, with significant implications for biosecurity and pandemic risk. Have you thought about dual use risks from your work and ways to mitigate that?
The recent Evo 2 paper from Arc Institute details out the red teaming work they did - we'll likely approach it in a similar way.
Why should we expect novel ML architectures to perform better than current methods like LASSO regression? If your task is disease risk prediction, this falls under "variance within the human distribution" which, as far as I know, should be explained mostly by additive effects of SNPs. Current missing heritability is more of an issue of insufficient data (like you say) and rare variants rather than an inefficiency of our models. Steve Hsu had a paper about why we should actually theoretically expect LASSO regression to be efficient here.
My impression is generally that genomics is one of the few areas in bio where fancy ML is ill-suited for the problem (specifically polygenic trait prediction). I'm curious if there's some premise I have that Tabula disagrees with that makes this venture promising.
Ammon here. We are in a data-limited environment where regularization is key. That's why LASSO regression has worked so well. Linearity itself (low parameter count) is regularization. Anything else (done naively) does not generalize. Of course, a (surprisingly) large amount of human variation does appear to be due to additive effects, and is thus well explained by linear models. But that's not the entire story. PRS models for many diseases still fall far short of broad-sense heritability. There is a gap to explain. The question, I think, comes does to the nature of that gap.
Talking to researchers, I encounter a few different theories: 1) rare SNPs not yet identified by GWAS, 2) structural variants not included in GWAS, 3) epistasis/dominance, 4) epigenetics, 5) gene/environment interactions (perhaps undermining the twin studies used to calculate broad-sense heritability. I'd love to hear other ideas.
To the extent that 2, 3, or 4 are true (for some important diseases) looking beyond current techniques seems necessary. If 1 is true, I still think there's lift from new approaches. Take a look at GPN-MSA. Reducing the size of the search space (calculating a prior over SNP pathogenicity from unsupervised methods) finds more loci.
I don't know what they have in mind, and I agree the first obvious thing to do is just get more data and try linear models. But there's plenty of reason to expect gains from nonlinear models, since broadsense heritability is higher than narrowsense, and due to missing heritability (though maybe it ought to be missing given our datasets), and due to theoretical reasons (quite plausibly there's multiple factors, as straw!tailcalled has described, e.g. in an OR of ANDs circuit; and generally nonlinearities, e.g. U-shaped responses in latents like "how many neurons to grow").
My guess, without knowing much, is that one of the first sorts of things to try is small circuits. A deep neural net (i.e. differentiable circuit) is a big circuit; it has many many hidden nodes (latent variables). A linear model is a tiny circuit: it has, say, one latent (the linear thing), maybe with a nonlinearity applied to that latent. (We're not counting the input nodes.)
What about small but not tiny circuits? You could have, for example, a sum of ten lognormals, or a product of ten sums. At a guess, maybe this sort of thing
- Captures substantially more of the structure of the trait, and so has in its hypothesis space predictors that are significantly better than any possible linear PGS;
- is still pretty low complexity / doesn't have vanishing gradients, or something--such that you can realistically learn given the fairly small datasets we have.
I've been looking at non-Euclidean loss functions in my PhD, and particularly at ultrametric loss functions. That gives you a very different kind of supervised learning (even if you are just doing linear regression). And it may be relevant here because it's very good at modelling hierarchies (e.g. ancestry).
So if you interpret 'different kind of architecture' as 'we need to do something other than what we're doing at the moment with Euclidean-based linear regression' then I agree with the post, but if it's 'we must do deep learning with neural networks' then I agree with Yejun.
ML arguments can take more data as input. In particular, the genomic sequence is not a predictor used in LASSO regression models: the variants are just arbitrarily coded as 0,1, or 2 alternative allele count. The LASSO models have limited ability to pool information across variants or across data modes. ML models like this one can (in theory) predict effects of variants just based off their sequence on data like RNA-sequencing (which shows which genes are actively being transcribed). That information is effectively pooled across variants and ties genomic sequence to another data type (RNA-seq). If you include that information into a disease-effect prediction model, you might improve upon the LASSO regression model. There are a lot of papers claiming to do that now, for example the BRCA1 supervised experiment in the EVO-2 paper. Of course, a supervised disease-effect prediction layer could be LASSO itself and just include some additional features derived from the ML model.
My take on missing heritability is summed up in Heritability: Five Battles, especially §4.3-4.4. Mental health and personality have way more missing heritability than things like height and blood pressure. I think for things like height and blood pressure etc., you’re limited by sample sizes and noise, and by SNP arrays not capturing things like copy number variation. Harris et al. 2024 says that there exist methods to extract CNVs from SNP data, but that they’re not widely used in practice today. My vote would be to try things like that, to try to squeeze a bit more predictive power in the cases like height and blood pressure where the predictors are already pretty good.
On the other hand, for mental health and personality, there’s way more missing heritability, and I think the explanation is non-additivity. I humbly suggest my §4.3.3 model as a good way to think about what’s going on.
If I were to make one concrete research suggestion, it would be: try a model where there are 2 (or 3 or whatever) latent schizophrenia subtypes. So then your modeling task is to jointly (1) assign each schizophrenic patient to one of the 2 (or 3 or whatever) latent subtypes, and (2) make a simple linear SNP predictor for each subtype. I’m not sure if anyone has tried this already, and I don’t personally know how to solve that joint optimization problem, but it seems like the kind of problem that a statistics-savvy person or team should be able to solve.
I do definitely think there are multiple disjoint root causes for schizophrenia, as evidenced for example by the fact that some people get the positive symptoms without the cognitive symptoms, IIUC. I have opinions (1,2) about exactly what those disjoint root causes are, but maybe that’s not worth getting into here. Ditto with autism having multiple disjoint root causes—for example, I have a kid who got an autism diagnosis despite having no sensory sensitivities, i.e. the most central symptom of autism!! Ditto with extroversion, neuroticism, etc. having multiple disjoint root causes, IMO.
Good luck! :)
Note that my suggestion (“…try a model where there are 2 (or 3 or whatever) latent schizophrenia subtypes. So then your modeling task is to jointly (1) assign each schizophrenic patient to one of the 2 (or 3 or whatever) latent subtypes, and (2) make a simple linear SNP predictor for each subtype…”)
…is a special case of @TsviBT’s suggestion (“what about small but not tiny circuits?”).
Namely, my suggestion is the case of the following “small but not tiny circuit”: X OR Y […maybe OR Z etc.].
This OR circuit is nice in that it’s a step towards better approximation almost no matter what the true underlying structure is. For example, if there’s a U-shaped quadratic dependency, the OR can capture whether you’re on the descending vs ascending side of the U. Or if there’s a sum of two lognormals, one is often much bigger than the other, and the OR can capture which one it is. Or whatever.
Thinking about it more, I guess the word “disjoint” in “disjoint root causes” in my comment is not quite right for schizophrenia and most other cases. For what little it’s worth, here’s the picture that was in my head in regards to schizophrenia:

The details don’t matter too much but see 1,2. The blue blob is a schizophrenia diagnosis. The purple arrows represent some genetic variant that makes cortical pyramidal neurons generally less active. For someone predisposed to schizophrenia mainly due to “unusually trigger-happy 5PT cortical neurons”, that genetic variant would be protective against schizophrenia. For someone predisposed to schizophrenia mainly due to “deficient cortex-to-cortex communication”, the same genetic variant would be a risk factor.
The X OR Y model would work pretty well for this—it would basically pull apart the people towards the top from the people towards the right. But I shouldn’t have said “disjoint root causes” because someone can be in the top-right corner with both contributory factors at once.
(I’m very very far from a schizophrenia expert and haven’t thought this through too much. Maybe think of it as a slightly imaginary illustrative example instead of a confident claim about how schizophrenia definitely works.)
Inspired by critical remarks from @Laura-2 about "bio/acc", my question is, when and how does something like this give rise to causal explanation and actual cures? Maybe GWAS is a precedent. You end up with evidence that a particular gene or allele is correlated with a particular trait, but you have no idea why. That lets you (and/or society) know some risks, but it doesn't actually eliminate disease, unless you think you can get there by editing out risky alleles, or just screening embryos. Otherwise this just seems to lead (optimistically) to better risk management, and (pessimistically) to a "Gattaca" society in which DNA is destiny, even more than it is now.
I'm no biologist. I'm hoping someone who is, can give me an idea of how far this GWAS-like study of genotype-phenotype correlations, actually gets us towards new explanations and new cures. What's the methodology for closing that gap? What extra steps are needed? How much have we benefited from GWAS so far?
Dear people at Tabula Bio,
I have a specific approach involving analytic combinatorics (see https://github.com/fargolo/AnalyticComb.jl/ ).
I recently developed it for dynamical systems successfully (recurrence analysis; see https://github.com/fargolo/SymbolicInference.jl ; https://doi.org/10.31219/osf.io/3ws85) and it seems particularly adequate for the underlying fuzzy and noisy genetic grammar.
See this work on cyphered manuscripts and related papers:
https://www.connectedpapers.com/main/2ac74c8b9ee318444a0ae5d4cf54fe909d8529a5/Decoding-Anagrammed-Texts-Written-in-an-Unknown-Language-and-Script/graph
I sent you an e-mail (at team@tabulabio.com) back in March (23rd), but received no response.
Best regards,
Felipe C. Argolo
Many of the most devastating human diseases are heritable. Whether an individual develops schizophrenia, obesity, diabetes, or autism depends more on their genes (and epi-genome) than it does on any other factor. However, current genetic models do not explain all of this heritability. Twin studies show that schizophrenia, for example, is 80% heritable (over a broad cohort of Americans), but our best genetic model only explains ~9% of variance in cases. To be fair, we selected a dramatic example here (models for other diseases perform far better). Still, the gap between heritability and prediction stands in the way of personalized genetic medicine.
We are launching Tabula Bio to close this gap. We have a three-part thesis on how to approach this.
We’re interested in probabilistic programming as a method to build such a model.
This is an ambitious idea, and we’re just getting started. But if we look into the future, to a world where humans have closed the heritability gap and personalized genetic medicine has eradicated great swaths of disease, it’s hard to imagine we did not get there via an effort like this.
Our team is currently Ammon Bartram, who was previously cofounder of Triplebyte and Socialcam, and Michael Poon, who has spent the past several years working on polygenic screening, an early employee at Twitch, and studied CS at MIT. We’re honored to have Michael Seibel and Emmett Shear among our initial investors.
Please reach out to us at team@tabulabio.com to help us harden our hypotheses or collaborate together on this mission! We're especially interested in talking with ML engineers interested in genomics and bioinformaticians interested in ML.