One more question for your list: what industries have not been subject to this regulatory ratchet and why not?
I‘m thinking of insecure software, although others may be able to come up with more examples. Right now software vendors have no real incentive to ship secure code. If someone sells a connected fridge which any thirteen-year-old can recruit into their botnet, there’s no consequence for the vendor. If Microsoft ships code with bugs and Windows gets hacked worldwide, all that they suffer is embarrassment[1]. And this situation has stayed stable since the invention of software. Even after high-publicity bugs like Heartbleed or NotPetya, the industry hasn’t suffered the usual regulatory response of ’something must be done, this is something so we’re going to do it’.
You don’t have to start with a pre-approval model. You could write a law requiring all software to be ‘reasonably secure’, and create a 1906-FDA type policing agency that would punish insecure software after the fact. This seems like the first step of the regulatory ratchet, it would be feasible, but no one (in any country?) has done it and I don’t know why.
We’ve also had demonstrations in principle of the ability to hack highly-regulated objects like medical devices and autos, but still no ratchet even in regulated domains and I don’t understand why not.
My best explanation for this non-regulation is that nothing bad enough has happened. We haven’t had any high-profile safety incidents where someone has died, and that is what starts the ratchet. But we have had high-profile hacks, and at least one has taken down hospitals and killed someone in a fairly direct way, and I don’t even remember any journalists pushing for regulation. I notice that I’m confused.
Software is an example of an industry where the first step of the ratchet never happened. Are there any industries which got a first step like a policing-agency and then didn’t ratchet from there even after high-publicity disasters? Can we learn how to prevent or interrupt the regulatory ratchet?
[1] Bruce Schneier has been pushing a liability model as a possible solution for at least ten years, but nothing has changed.
Health inspections can get crazy at restaurants too. My partner worked in the industry and is full of stories about this. Cupcakes with butter in the frosting being required to be kept in the refrigerator until sold. It being illegal to prep food in a food cart, then transfer it to a food truck for catering, despite such transfers being legal when the food is cooked in a restaurant. It being impossible to certify storage units to keep extra ingredients. Requirements that food carts have clean water tanks attached even when they are directly hooked up to clean city water. Requirements to track how long food has been hot held despite this making no real difference for food safety.
In most cases, workers deal with this by lying to or withholding information from inspectors.
As I wrote in a comment on From Oversight To Overkill:
There is no problem you can't solve with another level of indirection. So the obvious solution: Regulate the regulators. Make the regulators prove that a regulation they make or enforce is not killing more people than it saves.
Though, alas, I don't think we will see that or that it would be feasible.
I like the idea of using insurance, but note that the way insurance usually works is that now the insurer is forcing you to manage risks and establish "reasonable" procedures. The up-side is that the insurer hopefully has a better understanding of what things need to be improved (what is actually reasonable) and which do not and that there is competition between insurers. But it can take some time.
Do futarchies (regulatory processes driven by prediction markets) have a role to play here? I think they probably do. Approver's incentives (usually, their job security) aren't currently controlled in proportion to the outcomes of their decisions, the institution disproportionately pays too much attention to downside and not enough to upside, right?
To make people weigh risks in proportion to the potential upside, you may need to make a person's incentives and degree of influence accountable to something like a market.
(I've designed a bit in this area. Venture Granters. I wont attempt to adapt this one to drug regulation, not my domain. I feel it can be done, but it's going to need a lot more moving parts and it's going to have to be led more by projections than end results, because the end results of drug approvals often take a long time to manifest, and are large and irreversible.)
Insurance contracts are regulatory processes driven by prediction markets. If you have liability plus required insurance then different insurance companies compete with regulations that you have to follow to be eligible for their insurance contracts and the insurance companies trade on various futures to manage their risks.
Yeah. So to paraphrase, you could have a system where you can release something but only if you can find someone who will sell you insurance, then if your drug gives people cancer on a 20 year time delay the insurer has to pay for the chemo, and more for the damages that can't be repaired.
How do these systems address the fact that companies and their human executives often just wont viscerally care about consequences that're 20 years away? Or that sometimes the downsides will be so adverse that one org wont really be able to weigh them in proportion to the upsides via monetary incentives? (you can't punish an org with anything worse than bankruptcy, but sometimes the downsides are so bad that you need to.) With venture granters I tried to think of ways of increasing accountability beyond what bankruptcy allows. One example is prison sentences for insurers who can't pay liabilities, regardless of whether their mistakes were malicious. Not sure why we don't have that already. Another is network structures between insurers: they can make agreements to share liabilities to increase their 'load rating', but now they're going to have formalized incentives to audit each other, which I've heard via development economist Mushtaq Khan ( https://80000hours.org/podcast/episodes/mushtaq-khan-institutional-economics/ ) is a great way to keep an industry straight and to get the industry to participate fruitfully in generating effective regulation.
Clarify "Compete with regulations"?
We don't have many problems with insurance companies going broke and not being able to pay out.
The bigger problem is that "Ask a jury of your peers whether or not the drug was responsible for the cancer" is a crappy way to determine causality.
Clarify "Compete with regulations"?
Different insurance companies have different policies about what the buyer of the insurance company has to do to be eligible for them. Those policies are regulations.
We don't have many problems with insurance companies going broke and not being able to pay out.
How do you know this? (I'd be surprised if this was true, unless it was a result of a) insurance companies' contracts maintaining a bottomless barrel of excuses to not provide the service they sold, which is very common b) a result of forgiveness and bailouts which should not have happened or c) it basically does happen, but the brand gets sold to new owners and most of the staff and facilities are kept and no one notices. In which case I wouldn't be surprised at all, but I'm still right, these are all very problematic phenomena.)
Different insurance companies have different policies about what the buyer of the insurance company has to do to be eligible for them. Those policies are regulations.
That makes sense, so the government sets some low-level requirements that're risky to take on, simple, and abstract, something like, idk, "you'll be destroyed if The Weighers find that your approvals produce more harm than good" or something, and the insurance company builds specific instructions on top of that like "nothing bad will happen to you as long as you do these specific things"?
How do you know this?
It's the sort of problem that's newsworthy. If we would have many problems like that, people would be angry about it.
the insurance company builds specific instructions on top of that like "nothing bad will happen to you as long as you do these specific things"?
Or at least the amount of bad that happens is manageable for the insurance company.
If you look at malpractice insurance, some insurance providers require Board certification of doctors or give those that have it better rates.
Insurance is exactly a mechanism that transforms high-variance penalties in the future into consistent penalties in the present: the more risky you are, the higher your premiums.
Then insurance as you've defined it is not a specific mechanism, it's a category of mechanisms, most of which don't do the thing they're supposed to do. I want to do mechanism design. I want to make a proposal specific enough to be carried out irl.
I’m not suggesting that that liability law is the solution to everything. I just want to point out that other models exist, and sometimes they have even worked.
I don't understand the line of logic here. Showing that other models exist and finding a few examples where they worked isn't a compelling reason.
To change would require that the new model works significantly more often, with all the same real world conditions as the status-quo model.
If their success rate is roughly the same, or even worse, then I can't see any reason to change.
To change would require that the new model works significantly more often, with all the same real world conditions as the status-quo model.
Isn't your position infinitely conservative? If you have to demonstrate the efficacy of an alternative to that standard before you're allowed to try the alternative, then you can't ever try any alternatives.
Or put differently, why demand more from the alternative than from the status quo? We could instead demand that the status quo (e.g. the FDA's ultra-slow drug approval process) justify itself, and when it can't (as per the post above), replace it with whatever seems like the best idea at the time.
Isn't your position infinitely conservative? If you have to demonstrate the efficacy of an alternative to that standard before you're allowed to try the alternative, then you can't ever try any alternatives.
Did you misread? I wasn't commenting at all on experimentation.
To change would require that the new model works significantly more often, with all the same real world conditions as the status-quo model.
I don't think I misread, but I'll admit I don't understand. My point was that the quoted requirement sounds like it would make it impossible to ever replace or reform an entrenched paradigm like the review-and-approval model, or an entrenched institution like the FDA.
Because the only way to fulfill the requirement is to demonstrate that someone has already found a better solution and been allowed to implement it, which this requirement would forbid. Even if you're allowed to experiment, the requirement sounds too stringent to ever admit experimental evidence as sufficient.
Even looking at the case of 'replacing' or 'reforming' the FDA entirely, thankfully there is more than one authority, or country, in the world? And no one has a monopoly over humankind?
I'm not really sure how to explain this better, but here's a try:
It's clearly possible for some organizational structure, better then the FDA circa 2023, to come into existence at some point in the future and demonstrate that with concrete evidence and so on.
Of course it's theoretically possible for a supermajority of folks in the US to keep on ignoring all of it, but if that happens too many times then the US will be outcompeted and cease to exist eventually. So there's a self-correction dynamic built in for humankind, even in the most extreme of scenarios.
I've thought about an approach to this I call meta-regulation: regulations on what kind of regulations can be passed.
One of my favorite ideas is to limit the number of total regulations a given agency can set (or perhaps the total wordcount of its regulations, just to punish any gaming of this meta-regulation) to half of whatever it currently is. That way, once regulations are reduced to their newly declared peak, whenever regulators want to add a new regulation, they are forced to get rid of an existing one.
Hence they will be forced to do some form of cost-benefit analysis on the regulations they keep.
If we would make drug manufacturers liable for all health effects that people who take the drugs experience after taking the drugs, nobody would sell any drugs.
For vaccines, we have laws that explicitly remove liability from the producers of vaccines, because we believe that dragging vaccine manufacturers before a jury of people without any scientific training does not lead to good outcomes.
The lyme vaccine passed regulatory approval but liability concerns took LYMErix from the market.
Making OpenAI liable for everything that their model does would make them radically reduce the usage of the model. It's a strong move to slow down AI progress. It might even be stronger than the effect that IRB's have on slowing down science in general.
Right, and as Tyler Cowen pointed out in the article I linked to, we don't hold the phone company liable if, e.g., criminals use the telephone to plan and execute a crime.
So even if/when liability is the (or part of the) solution, it's not simple/obvious how to apply it. Needs good, careful thinking each time of where the liability should exist under what circumstances, etc. This is why we need experts in the law thinking about these things.
I have the impression that your post asserts that there's a problem with review-and-approval paradigm is in some way more problematic than other paradigms of how to regulate. It seems to me unclear why that would be true.
Right, and as Tyler Cowen pointed out in the article I linked to, we don't hold the phone company liable if, e.g., criminals use the telephone to plan and execute a crime.
While it sounds absurd to talk about this, there are legal proposals to do that at least for some crimes. In the EU there's the idea that there should be machine learning run on devices to detect when the user engages in some crimes and alerts authorities.
Brazil discusses at the moment legal responsibility for social media companies who publish "fake news".
I think the Nuclear Regulatory Commission (NRC) furnishes a clear case of this. In the 1960s, nuclear power was on a growth trajectory to provide roughly 100% of today’s world electricity usage. Instead, it plateaued at about 10%. The proximal cause is that nuclear power plant construction became slow and expensive, which made nuclear energy expensive, which mostly priced it out of the market. The cause of those cost increases is controversial, but in my opinion, and that of many other commenters, it was primarily driven by a turbulent and rapidly escalating regulatory environment around the late ‘60s and early ‘70s.
I do not follow you here. The paper you link to (in page 14) compares the energy generated by fuel types in three scenarios: what actually happened, and two contrefactual scenarios regarding how nuclear could have grown, one "linear" and one "accelerating".
In the actual scenario, nuclear indeed plateaued at about 10%. But in the "linear", it only reaches 25%, and even in the "accelerating", it only reaches about 75% of total capacity.
Did you take the most favorable scenario, and make it look significantly shinier, for good measure?
Looking at the “accelerating projection of 1960–1976” data points here, it reaches almost 3 TW by the mid-2010s:
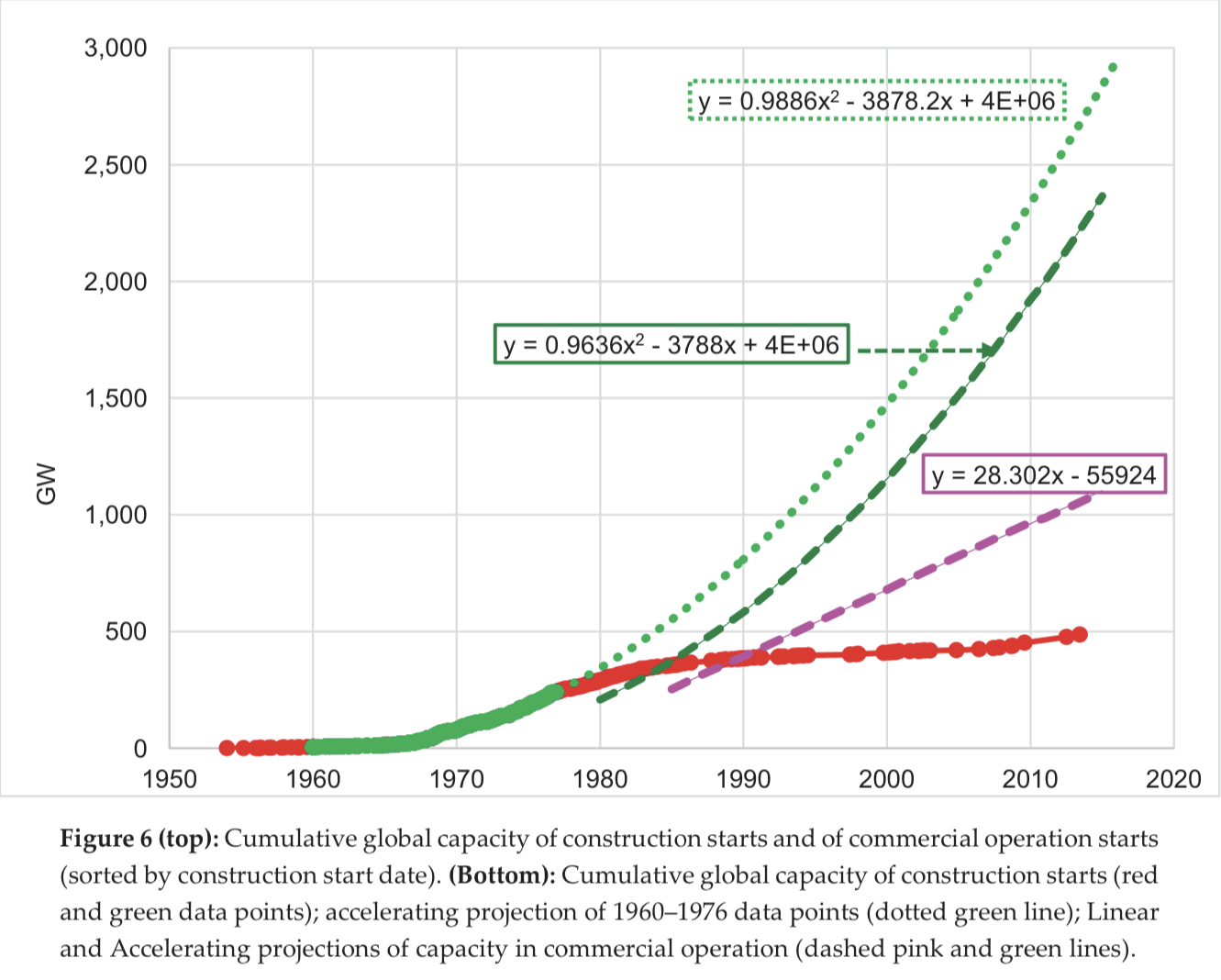
According to Our World in Data's energy data explorer, world electricity generation in 2021 was 27,812.74 TWh, which is 3.17 TW (using 1W = 8,766 Wh/year).
Comparing almost 3TW at about 2015 (just eyeballing the chart) to 3.17 TW in 2021, I say those are roughly equal. I did not make anything “significantly shinier”, or at least I did not intend to.
IRBs
Scott Alexander reviews a book about institutional review boards (IRBs), the panels that review the ethics of medical trials: From Oversight to Overkill, by Dr. Simon Whitney. From the title alone, you can see where this is going.
IRBs are supposed to (among other things) make sure patients are fully informed of the risks of a trial, so that they can give informed consent. They were created in the wake of some true ethical disasters, such as trials that injected patients with cancer cells (“to see what would happen”) or gave hepatitis to mentally defective children.
Around 1974, IRBs were instituted, and according to Whitney, for almost 25 years they worked well. The boards might be overprotective or annoying, but for the most part they were thoughtful and reasonable.
Then in 1998, during in an asthma study at Johns Hopkins, a patient died. Congress put pressure on the head of the Office for Protection from Research Risks, who overreacted and shut down every study at Johns Hopkins, along with studies at “a dozen or so other leading research centers, often for trivial infractions.” Some thousands of studies were ruined, costing millions of dollars:
Today IRB oversight has become, well, overkill. For one study testing the transfer of skin bacteria, the IRB thought that the consent form should warn patients of risks from AIDS (which you can’t get by skin contact) and smallpox (which has been eradicated). For a study on heart attacks, the IRB wanted patients—who are in the middle of a heart attack—to read and consent to a four-page form of “incomprehensible medicalese” listing all possible risks, even the most trivial. Scott’s review gives more examples, including his own personal experience.
In many cases, it’s not even as if a new treatment was being introduced: sometimes an existing practice (giving aspirin for a heart attack, giving questionnaires to psychology patients) was being evaluated for effectiveness. There was no requirement that patients consent to “risks” when treatment was given arbitrarily; but if outcomes were being systematically observed and recorded, the IRBs could intervene.
Scott summarizes the pros and cons of IRBs, including the cost of delayed treatments or procedure improvements:
FDA
The IRB story illustrates a common pattern:
The history of the FDA provides another example.
At the beginning of the 20th century, the drug industry was rife with shams and fraud. Drug ads made ridiculously exaggerated or completely fabricated claims: some claimed to cure consumption (that is, tuberculosis); another claimed to cure “dropsy and all diseases of the kidneys, bladder, and urinary organs”; another literally claimed to cure “every known ailment”. Many of these “drugs” contained no active ingredients, and turned out to be, for example, just cod-liver oil, or a weak solution of acid. Others contained alcohol—some in concentrations at the level of hard liquor, making patients drunk. Still others contains dangerous substances such as chloroform, opiates, or cocaine. Some of these drugs were marketed for use on children.
In 1906, in response to these and other problems, Congress passed the Pure Food & Drug Act, giving regulatory powers to what was then the USDA Bureau of Chemistry, and which would later become the FDA.
This did not look much like the modern FDA. It had no power to review new drugs or to approve them before they went on the market. It was more of a police agency, with the power to enforce the law after it had been violated. And the relevant law was mostly concerned with truth in advertising and labeling.
Then in 1937, the pharmaceutical company Massengill put a drug on the market called Elixir Sulfanilamide, one of the first antibiotics. The antibiotic itself was good, but in order to produce the drug in liquid form (as opposed to a tablet or powder), the “elixir” was prepared in a solution of diethylene glycol—which is a variant of antifreeze, and is toxic. Patients started dying. Massengill had not tested the preparation for toxicity before selling it, and when reports of deaths started to come in, they issued a vague recall without explaining the danger. When the FDA heard about the disaster, they forced Massengill to issue a clear warning, and then sent hundreds of field agents to talk to every pharmacy, doctor, and patient and track down every last vial of the poisonous drug, ultimately retrieving about 95% of what had been manufactured. Over 100 people died; if all of the manufactured drug had been consumed, it might have been over 4,000.
In the wake of this disaster, Congress passed the 1938 Food, Drug, and Cosmetic Act. This transformed the FDA from a police agency into a regulatory agency, giving them the power to review and approve all new drugs before they were sold. But the review process only required that drugs be shown safe; efficacy was not part of the review. Further, the law gave the FDA 60 days to reply to any drug application; if they failed to meet this deadline, then the drug was automatically approved.
I don’t know exactly how strict the FDA was after 1938, but the next fifteen years or so were the golden age of antibiotics, and during that period the mortality rate in the US decreased faster than at any other time in the 20th century. So if there was any overreach, it seems like it couldn’t have been too bad.
The modern FDA is the product of a different disaster. Thalidomide was a tranquilizer marketed to alleviate anxiety, trouble sleeping, and morning sickness. During toxicity testing, it seemed to be almost impossible to die from an overdose of thalidomide, which made it seem much safer than barbiturates, which were the main alternative at the time. But it was also promoted as being safe for pregnant mothers and their developing babies, even though no testing had been done to prove this. It turned out that when taken in the first several weeks of pregnancy, thalidomide caused horrible birth defects that resulted in deformed limbs and other organs, and often death. The drug was sold in Europe, where some 10,000 infants fell victim to it, but not in the US, where it was blocked by the FDA. Still, Americans felt they had had a close call, too close for comfort, and conditions were ripe for an overhaul of the law.
The 1962 Kefauver–Harris Amendment required, among other reforms, that new drugs be shown to be both safe and effective. It also lengthened the review period from 60 to 180 days, and if the FDA failed to respond in that time, drugs would no longer be automatically approved (in fact, it’s unclear to me what the review period even means anymore).
You might be wondering: why did a safety problem create an efficacy requirement in the law? The answer is a peek into how the sausage gets made. Senator Kefauver had been investigating drug pricing as early as 1959, and in the course of hearings, a former pharma exec remarked that some drugs on the market are not only overpriced, they don’t even work. This caught Kefauver’s attention, and in 1961 he introduced a bill that proposed enhanced controls over drug trials in order to ensure effectiveness. But the bill faced opposition, even from his own party and from the White House. When Kefauver heard about the thalidomide story in 1962, he gave it to the Washington Post, which ran it on the front page. By October, he was able to get his bill passed. So the law that was passed wasn’t even initially intended to address the crisis that got it passed.
I don’t know much about what happened in the ~60 years since Kefauver–Harris. But today, I think there is good evidence, both quantitative and anecdotal, that the FDA has become too strict and conservative in its approvals, adding needless delay that holds back treatments from patients. Scott Alexander tells the story of Omegaven, a nutritional fluid given to patients with digestive problems (often infants) that helped prevent liver disease: Omegaven took fourteen years to clear FDA’s hurdles, despite dramatic evidence of efficacy early on, and in that time “hundreds to thousands of babies … died preventable deaths.” Alex Tabarrok quotes a former FDA regulator saying:
Tabarrok also reports on a study that models the optimal tradeoff between approving bad drugs and failing to approve good drugs, and finds that “the FDA is far too conservative especially for severe diseases. FDA regulations may appear to be creating safe and effective drugs but they are also creating a deadly caution.” And Jack Scannell et al, in a well-known paper that coined the term “Eroom’s Law”, cite over-cautious regulation as one factor (out of four) contributing to ever-increasing R&D costs of drugs:
FDA delay was particularly costly during the covid pandemic. To quote Tabarrok again:
In short, an agency that began in order to fight outright fraud in a corrupt pharmaceutical industry, and once sent field agents on a heroic investigation to track down dangerous poisons, now displays an overly conservative, bureaucratic mindset that delays lifesaving tests and treatments.
NEPA
One element in common to all stories of regulatory overreach is the ratchet: once regulations are put in place, they are very hard to undo, even if they turn out to be mistakes, because undoing them looks like not caring about safety. Sometimes regulations ratchet up after disasters, as in the case of IRBs and the FDA. But they can also ratchet up through litigation. This was the case with NEPA, the National Environmental Policy Act.
Eli Dourado has a good history of NEPA. The key paragraph of the law requires that all federal agencies, in any “major action” that will significantly affect “the human environment,” must produce a “detailed statement” on the those effects, now known as an Environmental Impact Statement (EIS). In the early days, those statements were “less than ten typewritten pages,” but since then, “EISs have ballooned.”
In brief, NEPA allowed anyone who wanted to obstruct a federal action to sue the agency for creating an insufficiently detailed EIS. Each time an agency lost a case, it set a new precedent and increased the standard that all future EISes had to follow. Eli recounts how the word “major” was read out of the law, such that even minor actions required an EIS; the word “human” was read out of the law, interpreting it to apply to the entire environment; etc.
Eli summarizes:
Alec Stapp documents how NEPA has now become a barrier to affordable housing, transmission lines, semiconductor manufacturing, congestion pricing, and even offshore wind.
NRC
The problem with regulatory agencies is not that the people working there are evil—they are not. The problem is the incentive structure:
All of the incentives point in a single direction: towards more stringent regulations. No one regulates the regulators. This is the reason for the ratchet.
I think the Nuclear Regulatory Commission (NRC) furnishes a clear case of this. In the 1960s, nuclear power was on a growth trajectory to provide roughly 100% of today’s world electricity usage. Instead, it plateaued at about 10%. The proximal cause is that nuclear power plant construction became slow and expensive, which made nuclear energy expensive, which mostly priced it out of the market. The cause of those cost increases is controversial, but in my opinion, and that of many other commenters, it was primarily driven by a turbulent and rapidly escalating regulatory environment around the late ‘60s and early ‘70s.
At a certain point, the NRC formally adopted a policy that reflects the one-sided incentives: ALARA, under which exposure to radiation needs to be kept, not below some defined threshold of safety, but “As Low As Reasonably Achievable.” As I wrote in my review of Why Nuclear Power Has Been a Flop:
ALARA isn’t the singular root cause of nuclear’s problems (as Brian Potter points out, other countries and even the US Navy have formally adopted ALARA, and some of them manage to interpret “reasonable” more, well, reasonably). But it perfectly illustrates the problem. The one-sided incentives mean that regulators do not have to make any serious cost-benefit tradeoffs. IRBs and the FDA don’t pay a price for the lives lost while trials or treatments are waiting on approval. The EPA (which now reviews environmental impact statements) doesn’t pay a price for delaying critical infrastructure. And the NRC doesn’t pay a price for preventing the development of abundant, cheap, reliable, clean energy.
Google
All of these examples are government regulations, but a similar process happens inside most corporations as they grow. Small startups, hungry and having nothing to lose, move rapidly with little formal process. As they grow, they tend to add process, typically including one or more layers of review before products are launched or other decisions are made. It’s almost as if there is some law of organizational thermodynamics decreeing that bureaucratic complexity can only ever increase.
Praveen Seshadri was the co-founder of a startup that was acquired by Google. When he left three years later, he wrote an essay on “how a once-great company has slowly ceased to function”:
What Google has in common with a regulatory agency is that (according to Seshadri at least) its employees are driven by risk aversion:
A “minor change to a minor product” requires “literally 15+ approvals in a ‘launch’ process that mirrors the complexity of a NASA space launch,” any non-obvious decision is avoided because it “isn’t group think and conventional wisdom,” and everyone tries to placate everyone else up and down the management chain to avoid conflict.
A startup that operated this way would simply go out of business; Google can get away with this bureaucratic bloat because their core ads business is a cash cow that they can continue to milk, at least for now. But in general, this kind of corporate sclerosis leaves a company vulnerable to changes in technology and markets (as indeed Google seems to be falling behind startup competitors in AI).
The difference with regulation is that there is no requirement for agencies to serve customers in order to stay in existence, and no competition to disrupt their complacency, except at the international level. If you want to build a nuclear plant, you obey the NRC or you build outside the US.
Against the review-and-approval model
In the wake of disaster, or even in the face of risk, a common reaction is to add a review-and-approval process. But based on examples such as these, I now believe that the review-and-approval model is broken, and we should find better ways to manage risk and create safety.
Unfortunately, review-and-approval is so natural, and has become so common, that people often assume it is the only way to control or safeguard anything, as if the alternative is anarchy or chaos. But there are other approaches.
One example I have discussed is factory safety in the early 20th century, which was driven by a change to liability law. The new law made it easier for workers and their families to receive compensation for injury or death, and harder for companies to avoid that liability. This gave factories the legal and financial incentive to invest in safety engineering and to address the root causes of accidents in the work environment, which ultimately reduced injury rates by around 90%.
Jack Devanney has also discussed liability as part of a better scheme for nuclear power regulation. I have commented on liability in the context of AI risk, and Robin Hanson wrote an essay with a proposal (see however Tyler Cowen’s pushback on the idea). And Alex Tabarrok mentioned to me that liability appears to have driven remarkable improvements in anesthesiology.
I’m not suggesting that that liability law is the solution to everything. I just want to point out that other models exist, and sometimes they have even worked.
Open questions
Some things I’d like to learn more about:
Thanks to Tyler Cowen, Alex Tabarrok, Eli Dourado, and Heike Larson for commenting on a draft of this essay.