In the course of my life, there have been a handful of times I discovered an idea that changed the way I thought about where our species is headed. The first occurred when I picked up Nick Bostrom’s book “superintelligence” and realized that AI would utterly transform the world. The second was when I learned about embryo selection and how it could change future generations. And the third happened a few months ago when I read a message from a friend of mine on Discord about editing the genome of a living person.
We’ve had gene therapy to treat cancer and single gene disorders for decades. But the process involved in making such changes to the cells of a living person is excruciating and extremely expensive. CAR T-cell therapy, a treatment for certain types of cancer, requires the removal of white blood cells via IV, genetic modification of those cells outside the body, culturing of the modified cells, chemotherapy to kill off most of the remaining unmodified cells in the body, and reinjection of the genetically engineered ones. The price is $500,000 to $1,000,000.
And it only adds a single gene.
This is a big problem if you care about anything besides monogenic diseases. Most traits, like personality, diabetes risk, and intelligence are controlled by hundreds to tens of thousands of genes, each of which exerts a tiny influence. If you want to significantly affect someone’s risk of developing lung cancer, you probably need to make hundreds of edits.
If changing one gene to treat cancer requires the involved process described above, what kind of tortuous process would be required to modify hundreds?
It seemed impossible. So after first thinking of the idea well over a year ago, I shelved it without much consideration. My attention turned to what seemed like more practical ideas such as embryo selection.
Months later, when I read a message from my friend Kman on discord talking about gene editing adults in adult brains, I was fairly skeptical it was worth my time.
How could we get editing agents into all 200 billion brain cells? Wouldn’t it cause major issues if some cells received edits and others didn’t? What if the gene editing tool targeted the wrong gene? What if it provoked some kind of immune response?
But recent progress in AI had made me think we might not have much time left before AGI, so given that adult gene editing might have an impact on a much shorter time scale than embryo selection, I decided it was at least worth a look.
So I started reading. Kman and I pored over papers on base editors and prime editors and in-vivo delivery of CRISPR proteins via adeno-associated viruses trying to figure out whether this pipe dream was something more. And after a couple of months of work, I have become convinced that there are no known fundamental barriers that would prevent us from doing this.
There are still unknowns which need to be worked on. But many pieces of a viable editing protocol have already been demonstrated independently in one research paper or another.
If this works, it’s going to be a very big deal
It is hard to overstate just how big a deal it would be if we could edit thousands of genes in cells all over the human body. Nearly every trait and every disease you’ve ever heard of has a significant genetic component, from intelligence to breast cancer. And the amount of variance already present in the human gene pool is stunning. For many traits, we could take someone from the 3rd percentile to the 97th percentile by editing just 20% of the genes involved in determining that trait.
Tweaking even a dozen genes might be able to halt the progression of Alzheimer’s, or alleviate the symptoms of major autoimmune diseases.
The same could apply to other major causes of aging: diabetes, heart disease, cancers. All have genetic roots to some degree or other. And all of this could potentially be done in people who have already been born.
Of particular interest to me is whether we could modify intelligence. In the past ten thousand years, humans have progressed mostly through making changes to their tools and environment. But in that time, human genes and human brains have changed little. With limitations to brain power, new impactful ideas naturally become harder to find.
This is particularly concerning because we have a number of extremely important technical challenges facing us today and a very limited number of people with the ability to make progress on them. Some may simply be beyond the capabilities of current humans.
Of these, the one I think about most is technical AI alignment work. It is not an exaggeration to say that the lives of literally everyone depend on whether a few hundred engineers and mathematicians can figure out how to control the machines built by the mad scientists in the office next door.
Demis Hassabis has a famous phrase he uses to describe his master plan for Deepmind: solve intelligence, then use that intelligence to solve everything else. I have a slightly more meta level plan to make sure Demis’s plan doesn’t kill everyone; make alignment researchers smarter, then kindly ask those researchers to solve alignment.
This is quite a grandiose plan for someone writing a LessWrong blog post. And there’s a decent chance it won’t work out for either technical reasons or because I can't find the resources and talent to help me solve all the technical challenges. At the end of the day, whether or not anything I say has an impact will depend on whether I or someone else can develop a working protocol.
How does intelligence even work at a genetic level?
Our best estimate based on the last decade of data is that the genetic component of intelligence is controlled by somewhere between 10,000 and 24,000 variants. We also know that each one, on average, contributes about +-0.2 IQ points.
Genetically altering IQ is more or less about flipping a sufficient number of IQ-decreasing variants to their IQ-increasing counterparts. This sounds overly simplified, but it’s surprisingly accurate; most of the variance in the genome is linear in nature, by which I mean the effect of a gene doesn’t usually depend on which other genes are present.
So modeling a continuous trait like intelligence is actually extremely straightforward: you simply add the effects of the IQ-increasing alleles to to those of the IQ-decreasing alleles and then normalize the score relative to some reference group.
To simulate the effects of editing on intelligence, we’ve built a model based on summary statistics from UK Biobank, the assumptions behind which you can find in the appendix
Based on the model, we can come to a surprising conclusion: there is enough genetic variance in the human population to create a genome with a predicted IQ of about 900. I don’t expect such an IQ to actually result from flipping all IQ-decreasing alleles to their IQ-increasing variants for the same reason I don’t expect to reach the moon by climbing a very tall ladder; at some point, the simple linear model will break down.
But we have strong evidence that such models function quite well within the current human range, and likely somewhat beyond it. So we should actually be able to genetically engineer people with greater cognitive abilities than anyone who’s ever lived, and do so without necessarily making any great trade-offs.
Even if a majority of iq-increasing genetic variants had some tradeoff such as increasing disease risk (which current evidence suggests they mostly don’t), we could always select the subset that doesn’t produce such effects. After all, we have 800 IQ points worth of variants to choose from!
Total maximum gain
Given the current set of publicly available intelligence-associated variants available in UK biobank, here’s a graph showing the expected effect on IQ from editing genes in adults.
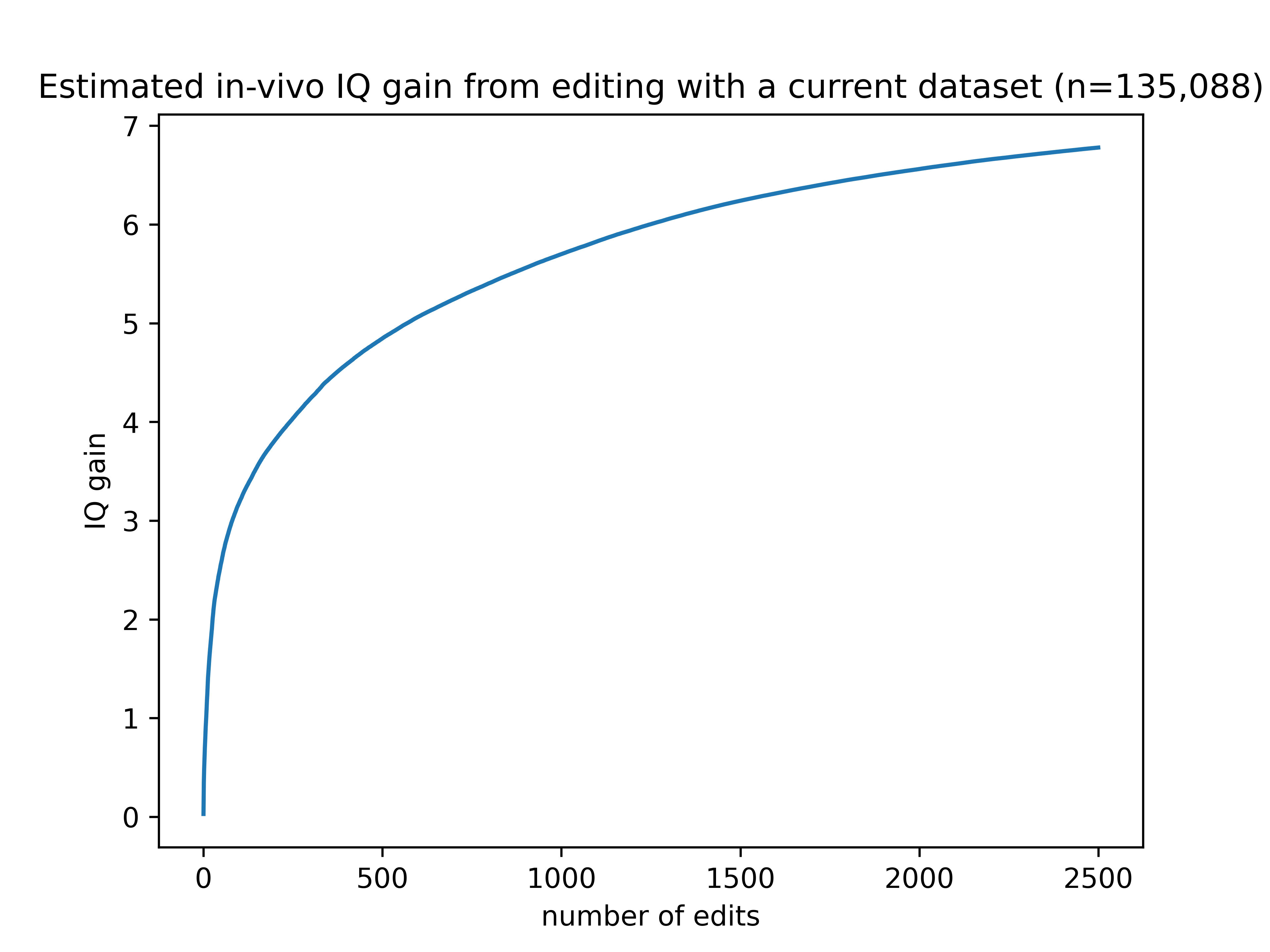
Not very impressive! There are several factors at play deflating the benefits shown by this graph.
- As a rough estimate, we expect about 50% of the targeted edits to be successfully made in an average brain cell. The actual amount could be more or less depending on editor and delivery efficiency.
- Some genes we know are involved in intelligence will have zero effect if edited in adults because they primarily influence brain development (see here for more details). Though there is substantial uncertainty here, we are assuming that, on average, an intelligence-affecting variant will have only 50% of the effect size as it would if edited in an embryo.
- We can only target about 90% of genetic variants with prime editors.
- About 98% of intelligence-affecting alleles are in non-protein-coding regions. The remaining 2% of variants are in protein-coding regions and are probably not safe to target with current editing tools due to the risk of frameshift mutations.
- Off-target edits, insertions and deletions will probably have some negative effect. As a very rough guess, we can probably maintain 95% of the benefit after the negative effects of these are taken into account.
- Most importantly, the current intelligence predictors only identify a small subset of intelligence-affecting genetic variants. With more data we can dramatically increase the benefit from editing for intelligence. The same goes for many other traits such as Alzheimer’s risk or depression.
The limitations in 1-5 together reduce the estimated effect size by 79% compared to making perfect edits in an embryo. If you could make those same edits in an embryo, the gains would top out around 30 IQ points.
But the bigger limitation originates from the size of the data set used to train our predictor. The more data used to train an intelligence predictor, the more of those 20,000 IQ-affecting variants we can identify, and the more certain we can be about exactly which variant among a cluster is actually causing the effect.
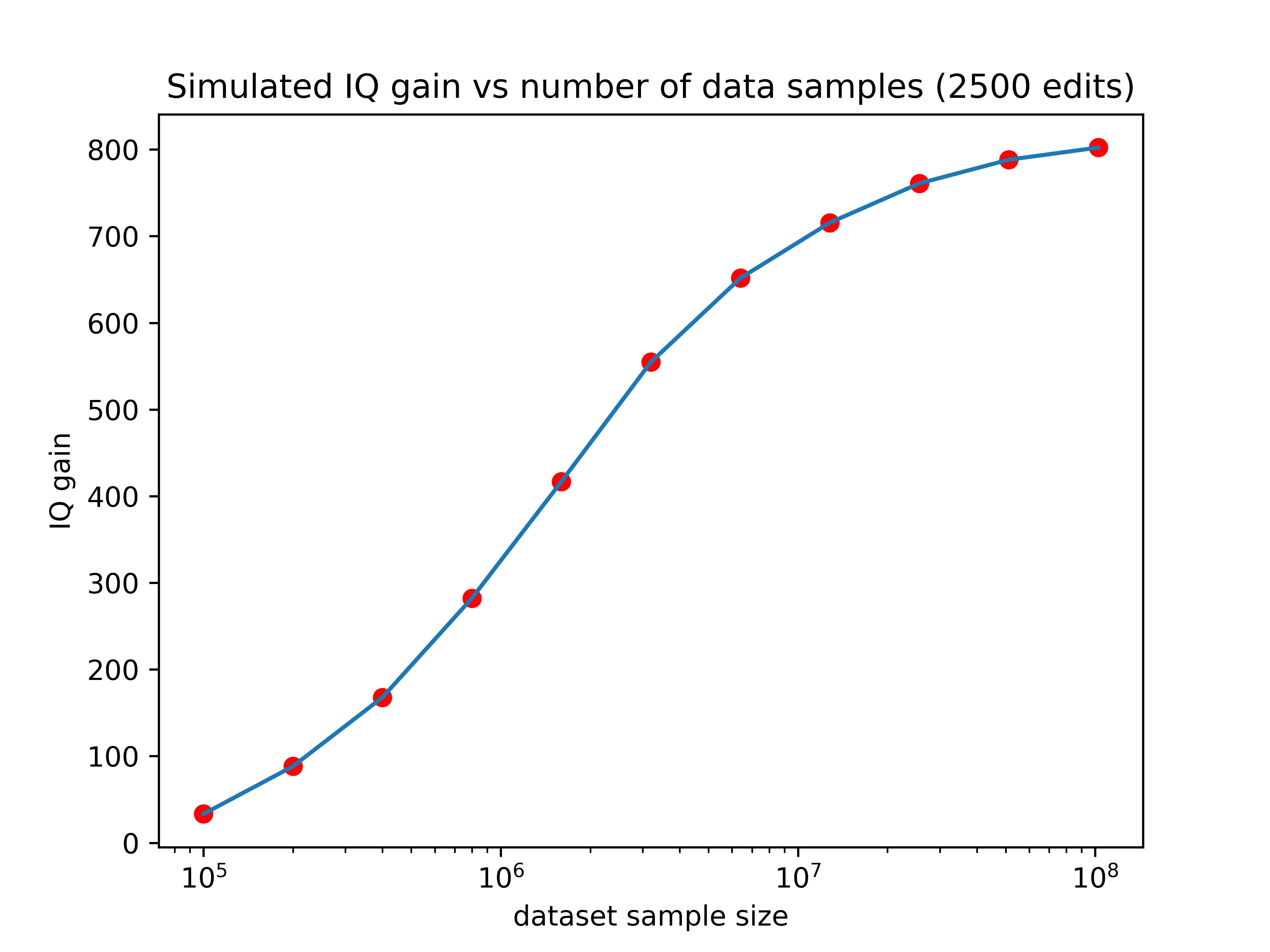
And the more edits you can make, the better you can take advantage of that additional data. You can see this demonstrated pretty clearly in the graph below, where each line represents a differently sized training set.
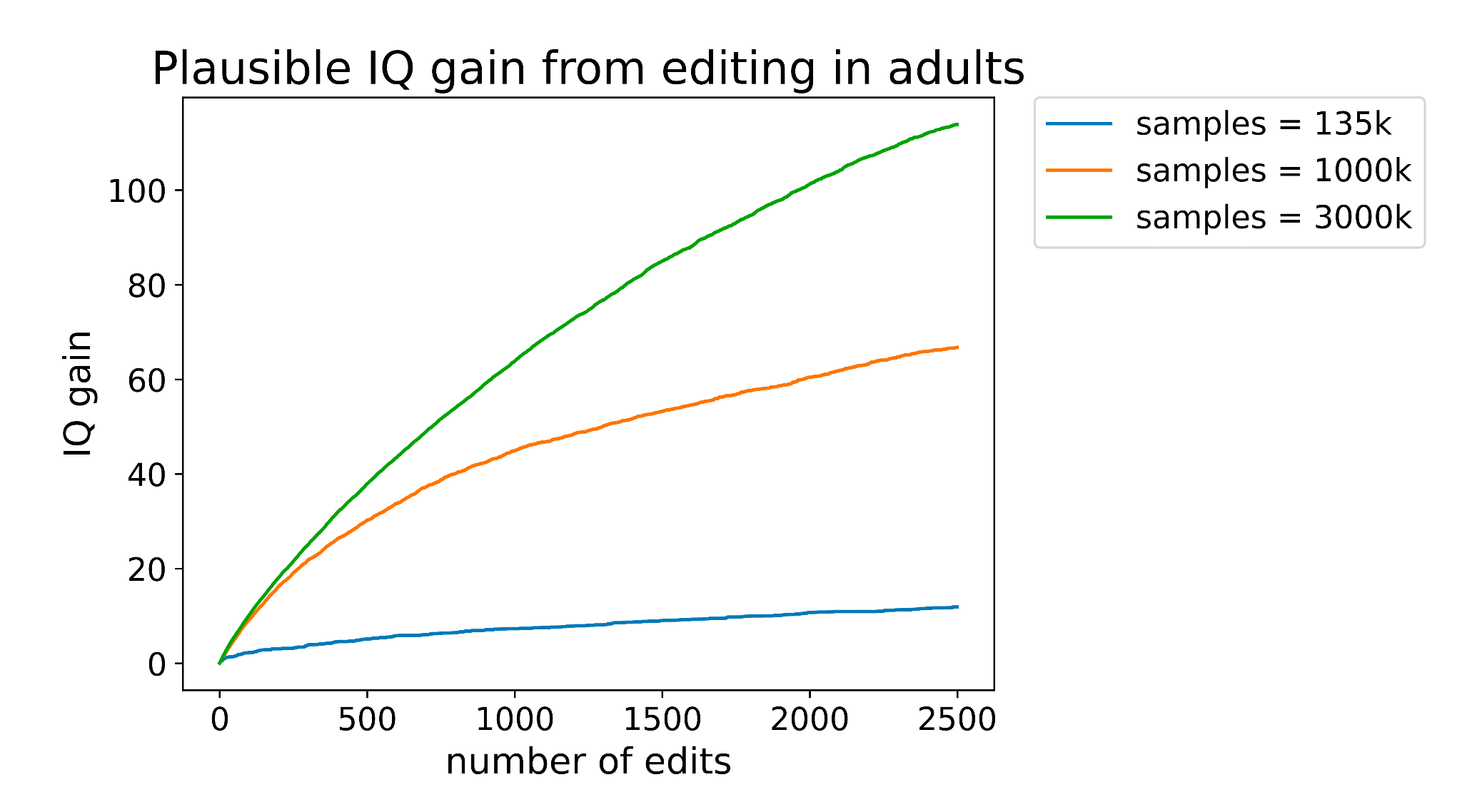
Our current predictors are trained using about 135,000 samples, which would place it just above the lowest line on the graph. There are existing databases right now such as the million veterans project with sample sizes of (you guessed it) one million. The expected gain from editing using a predictor trained on such a data set is shown by the orange line in the graph above.
Companies like 23&Me genotyped their 12 millionth customer two years ago and could probably get at perhaps 3 million customers to take an IQ test or submit SAT scores. A predictor trained with that amount of data would perform about as well as the green line on the graph above.
So larger datasets could increase the effect of editing by as much as 13x! If we hold the number of edits performed constant at 2500, here’s how the expected gain would vary as a function of training set size:
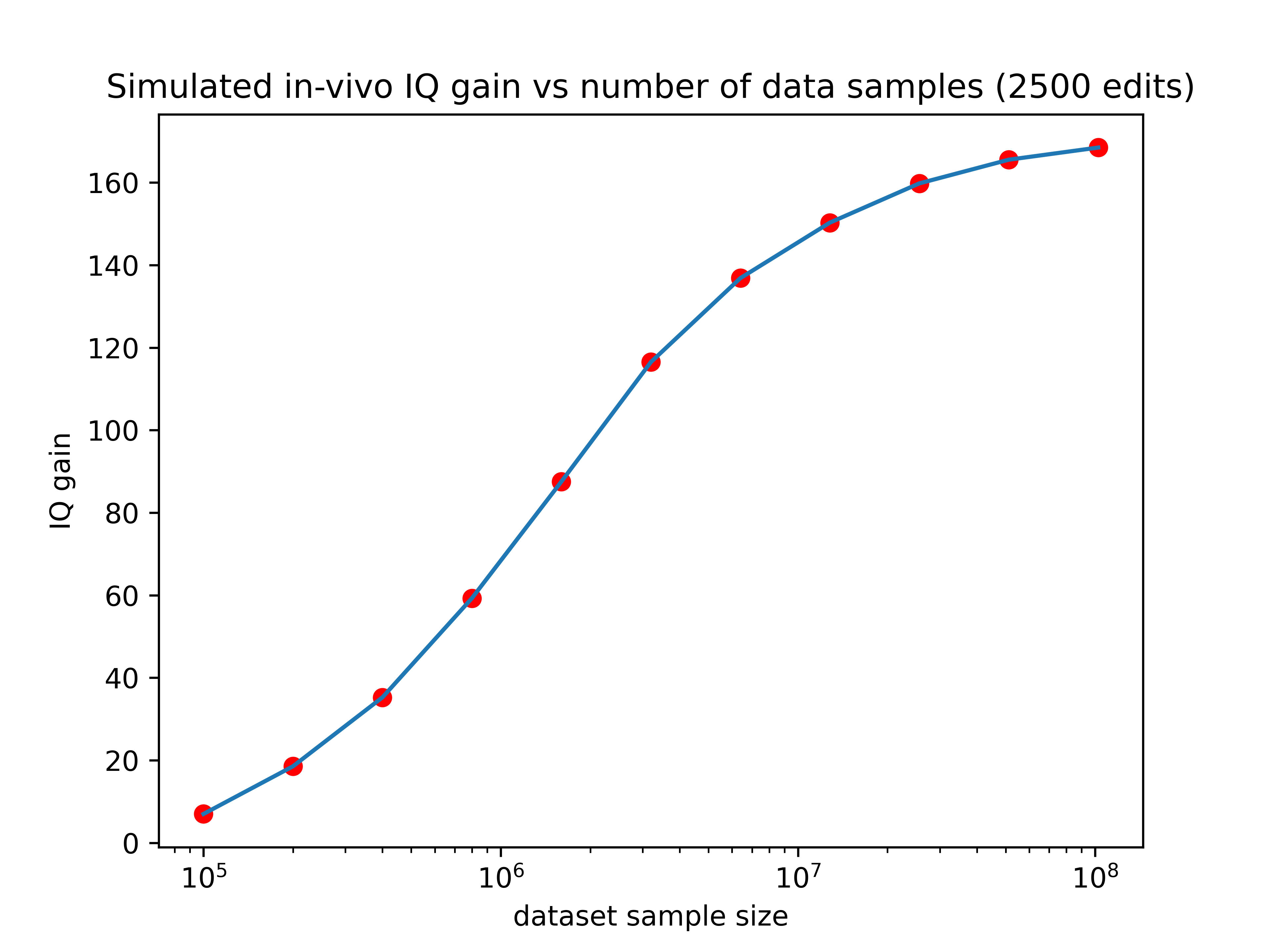
Now we’re talking! If someone made a training set with 3 million data points (a realistic possibility for companies like 23&Me), the IQ gain could plausibly be over 100 points (though keep in mind the uncertainties listed in the numbered list above). Even if we can only make 10% of that many edits, the expected effect size would still be over one standard deviation.
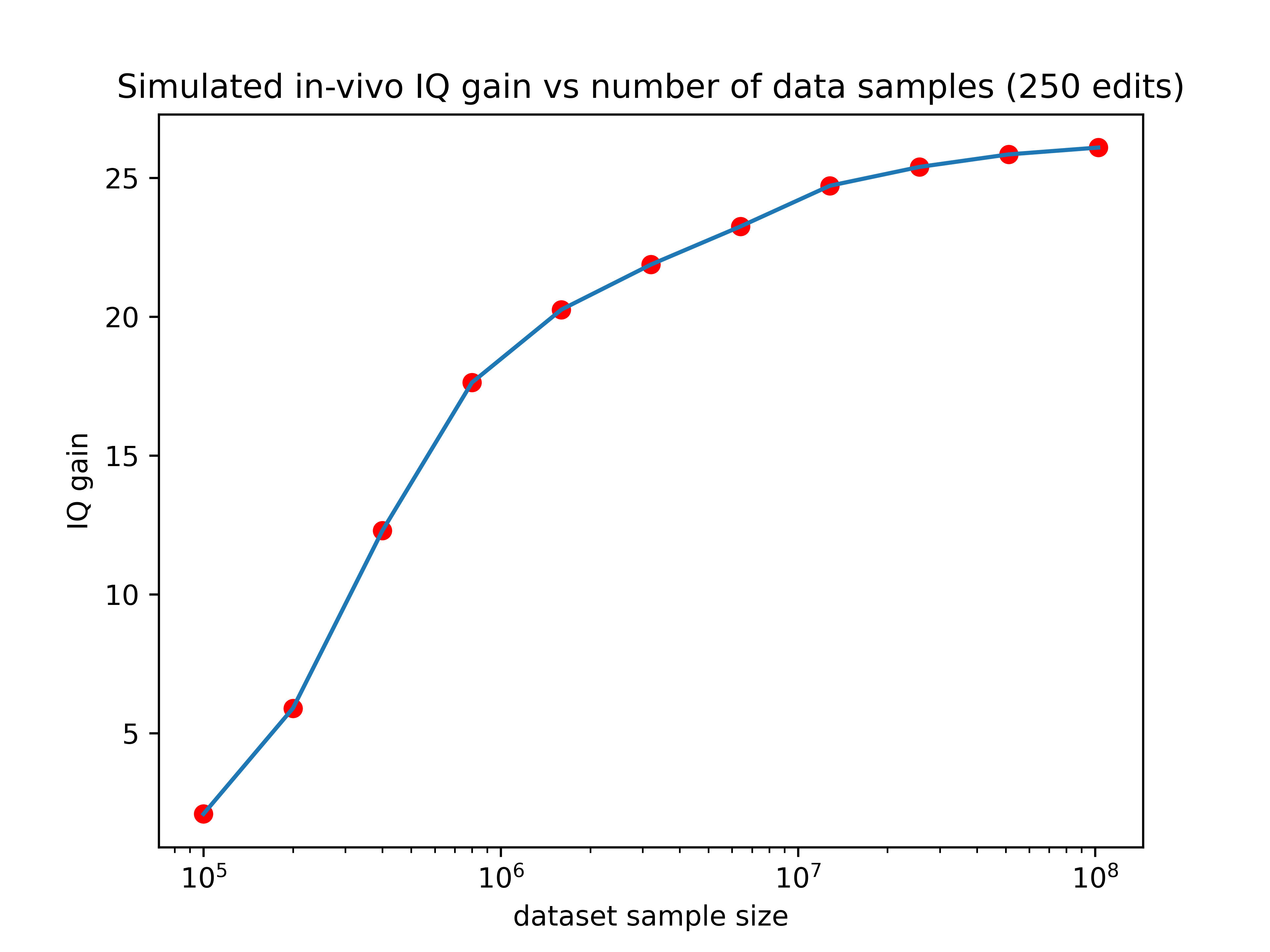
Lastly, just for fun, let’s show the expected effects of 2500 edits if they were made in an embryo and we didn’t have to worry about off-targets or limitations of editors (don’t take this graph too seriously)
The sample size issue could be solved pretty easily by any large government organization interested in collecting intelligence data on people they’ve already genotyped. However, most of them are not currently interested.
Why has it been so hard to get larger sample sizes?
There is a taboo against direct investigation of the genetics of intelligence in academia and government funding agencies. As a result, we have collected a pitifully small amount of data about intelligence, and our predictors are much worse as a result.
So people interested in intelligence instead have to research this proxy trait that is correlated with intelligence called “educational attainment”. Research into educational attainment IS allowed, so we have sample sizes of over 3 million genome samples with educational attainment labels and only about 130,000 for intelligence.

Government agencies have shown they could solve the data problem if they feel so inclined. As an alternative, if someone had a lot of money they could simply buy a direct-to-consumer genetic testing company and either directly ask participants to take an IQ test or ask them what their SAT scores were.
Or someone who wants to do this could just start their own biobank.
How can you even make these edits anyways? Isn’t CRISPR prone to errors?
Another of my initial doubts stemmed from my understanding of gene editing tools like CRISPR, and their tendency to edit the wrong target or even eliminate entire chromosomes.
You see, CRISPR was not designed to be a gene editing tool. It’s part of a bacterial immune system we discovered all the way back in the late 80s and only fully understood in 2012. The fact that it has become such a versatile gene editing tool is a combination of luck and good engineering.
The gene editing part of the CRISPR system is the Cas9 protein, which works like a pair of scissors. It chops DNA in half at a specific location determined by this neat little attachment called a “guide RNA”. Cas9 uses the guide RNA like a mugshot, bumping around the cell’s nucleus until it finds a particular three letter stretch of DNA called the PAM. Once it finds one, it unravels the DNA starting at the PAM and checks to see if the guide RNA forms a complementary match with the exposed DNA.
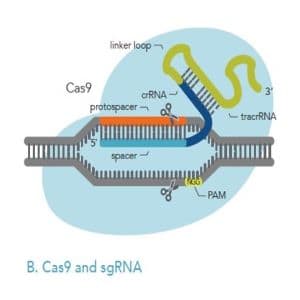
Cas9 can actually measure how well the guide RNA bonds to the DNA! If the hybridization is strong enough (meaning the base pairs on the guide RNA are complementary to those of the DNA), then the Cas9 “scissors” will cut the DNA a few base pairs to the right of the PAM.
So to replace a sequence of DNA, one must introduce two cuts: one at the start of the DNA and one at the end of it.
Once Cas9 has cut away the part one wishes removed, a repair template must be introduced into the cell. This template is used by the cell’s normal repair processes to fill in the gap.
Or at least, that’s how it’s supposed to work. In reality a significant portion of the time the DNA will be stitched back together wrong or not at all, resulting in death of the chromosome.
This is not good news for the cell.
So although CRISPR had an immediate impact on the life sciences after it was introduced in 2012, it was rightfully treated with skepticism in the context of human genome editing, where deleting a chromosome from someone’s genome could result in immediate cell death.
Had nothing changed, I would not be writing this post. But in 2016, a new type of CRISPR was created.
The first base editor
Many of the problems with CRISPR stem from the fact that it cuts BOTH strands of DNA when it makes an edit. A double stranded break is like a loud warning siren broadcast throughout the cell. It can result in a bunch of new base pairs being added or deleted at the break site. Sometimes it even results in the immune system ordering the cell to kill itself.
So when David Liu started thinking about how to modify CRISPR to make it a better gene editor, one of the first things he tried to do was prevent CRISPR from making double-stranded breaks.
He and members of his lab came up with a very creative way of doing this. They modified the Cas9 scissors so that they would still seek out and bind to a matching DNA sequence, but wouldn’t cut the DNA. They then attached a new enzyme to Cas9 called a “deaminase”. Its job is to change a single base through a chemical reaction.
They called this new version of CRISPR a “base editor”
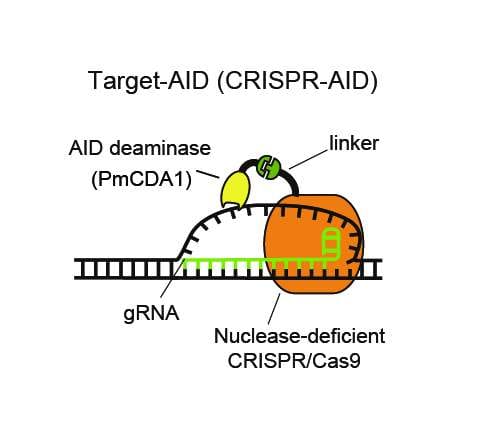
There’s a lot more detail to go into regarding how base editors work which I will explain in another post, but the most noteworthy trait of base editors is how reliable they are in comparison to old school Cas9.
Base editors make the correct edit between 60 and 180 times as often as the original Cas9 CRISPR. They also make 92% fewer off-target edits and 99.7% fewer insertion and deletion errors.
The most effective base editor I’ve found so far is the transformer base editor, which has an editing efficiency of 90% in T cells and no detected off-targets.
But despite these considerable strength, base editors still have two significant weaknesses:
- Of the 12 possible base pair swaps, they can only target 6.
- With some rare exceptions they can usually only edit 1 base pair at a time.
To address these issues, a few members of the Liu lab began working on a new project: prime editors.
Prime editors; the holy grail of gene editing technology?
Like base editors, prime editors only induce single stranded breaks in DNA, significantly reducing insertion and deletion errors relative to old school CRISPR systems. But unlike base editors, prime editors can change, insert, or delete up to about a hundred base pairs at a time.
The core insight leading to their creation was the discovery that the guide RNA could be extended to serve as a template for DNA modifications.
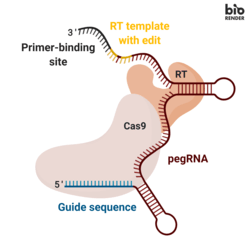
To greatly oversimplify, a prime editor cuts one of the DNA strands at the edit site and adds new bases to the end of it using an extension of the guide RNA as a template. This creates a bit of an awkward situation because now there are two loose flaps of DNA, neither of which are fully attached to the other strand.
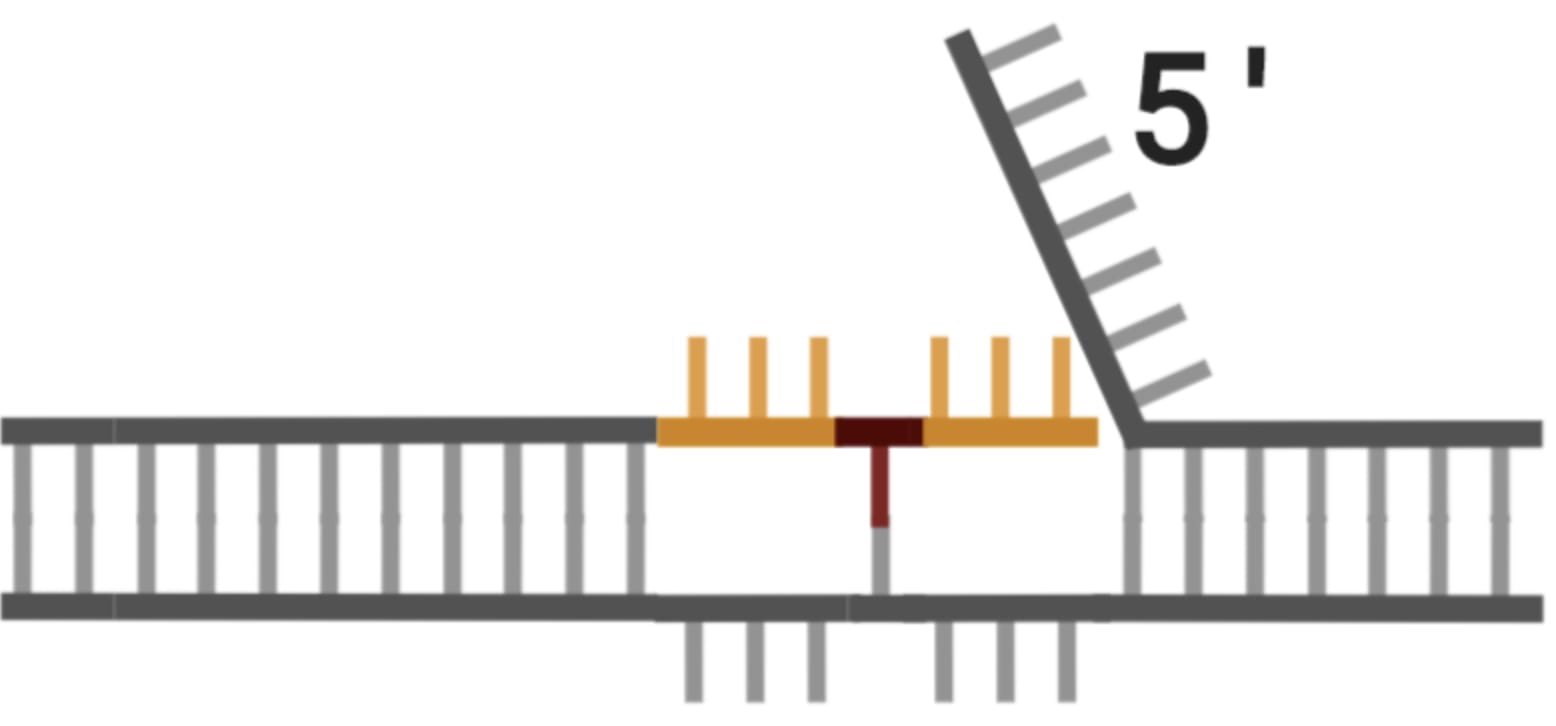
Thankfully the repair enzyme in charge of fixing these types of problems (which are a necessary part of normal cellular function), really likes to cut the unedited flap for reasons I won’t get into. Once it has cut away the other flap and stitched together the edited strand, the situation looks like this:
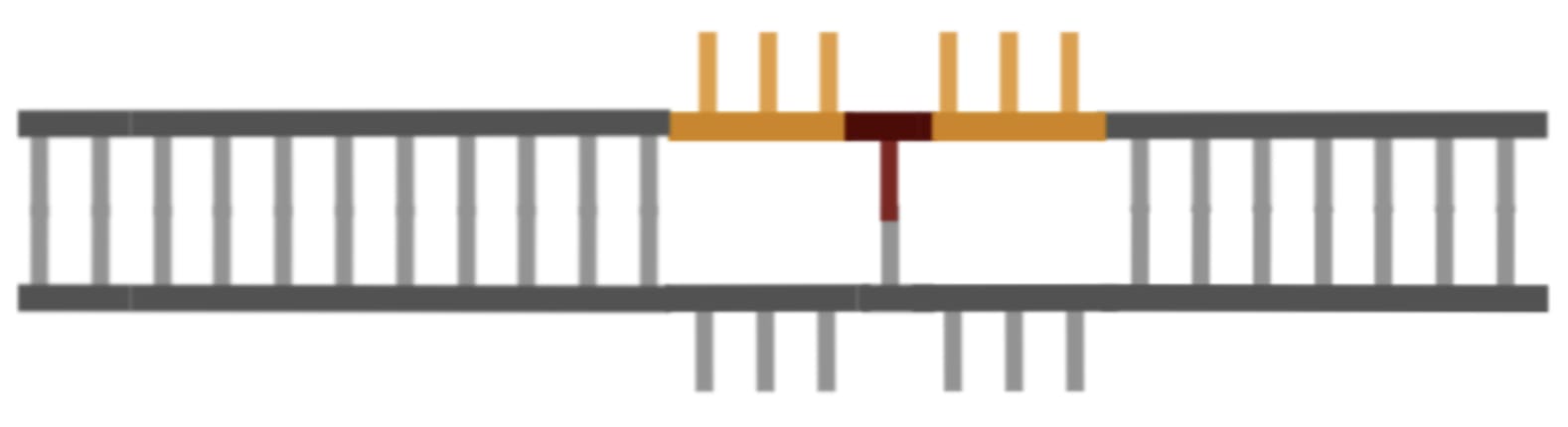
There’s still one problem left to fix though: the unedited strand is… well… unedited.
To fix this, Liu and Co use another enzyme to cut the bottom strand of DNA near the edit site, which tricks the repair enzymes in charge of fixing mismatches into thinking that the unedited strand has been damaged and needs to be fixed so that it matches the edited one.
So the cell’s own repair enzymes actually complete the edit for us, which is pretty neat if you ask me.
Together, prime editors and base editors give us the power to edit around 90% of the genetic variants we’re interested in with very high precision. Without them, editing genes in a living human body would be infeasible due to off-target changes.
How do you even get editors into brain cells in the first place?
There are roughly 200 billion cells in an average brain. You can’t just use a needle to inject a bunch of gene editing tools into each one. So how do we get them inside?
There are several options available to us including viral proteins, but the delivery platform I like the most is called a lipid nanoparticle. Let me explain the idea.
Dual use technology
Remember COVID vaccines? The first ones released used this new technology called “mRNA”. They worked a little differently from old school immunizations.
The goal of a vaccine is to expose your immune system to part of a virus so that it will recognize and kill it in the future.
One way to do this is by taking a little piece of the virus and injecting it into your body. This is how the Novavax vaccine worked.
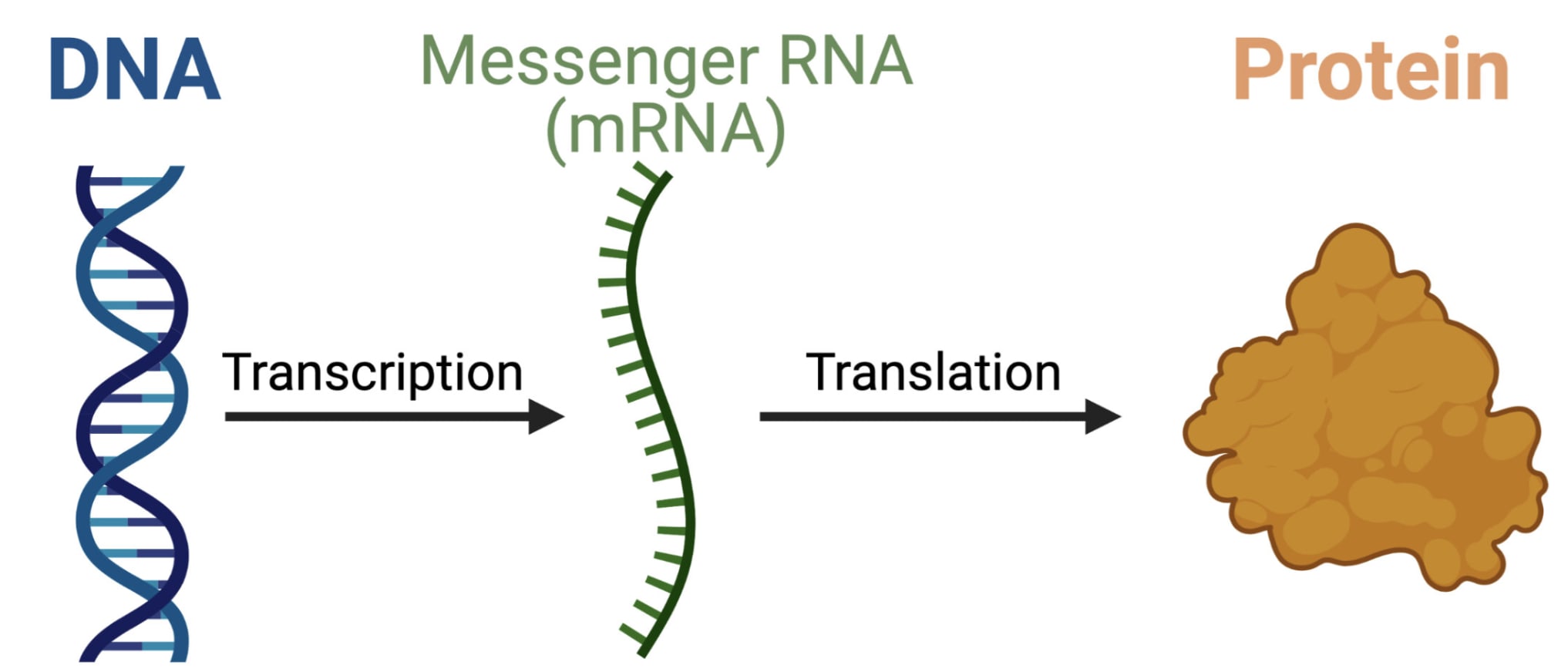
Another way is by sending an instruction to MAKE part of the virus to your body’s own cells. This is how the Pfizer and Moderna vaccines worked. And the instruction they used was a little piece of ribonucleic acid called mRNA.
mRNA is an instruction to your cells. If you can get it inside one, the cell’s ribosomes will transcribe mRNA into a protein. It’s a little bit like an executable file for biology.
By itself, mRNA is pretty delicate. The bloodstream is full of nuclease enzymes that will chop them into itty-bitty pieces.
Also, because mRNA basically has root level access to your cells, your body doesn’t just shuttle it around and deliver it like the postal service. That would be a major security hazard.
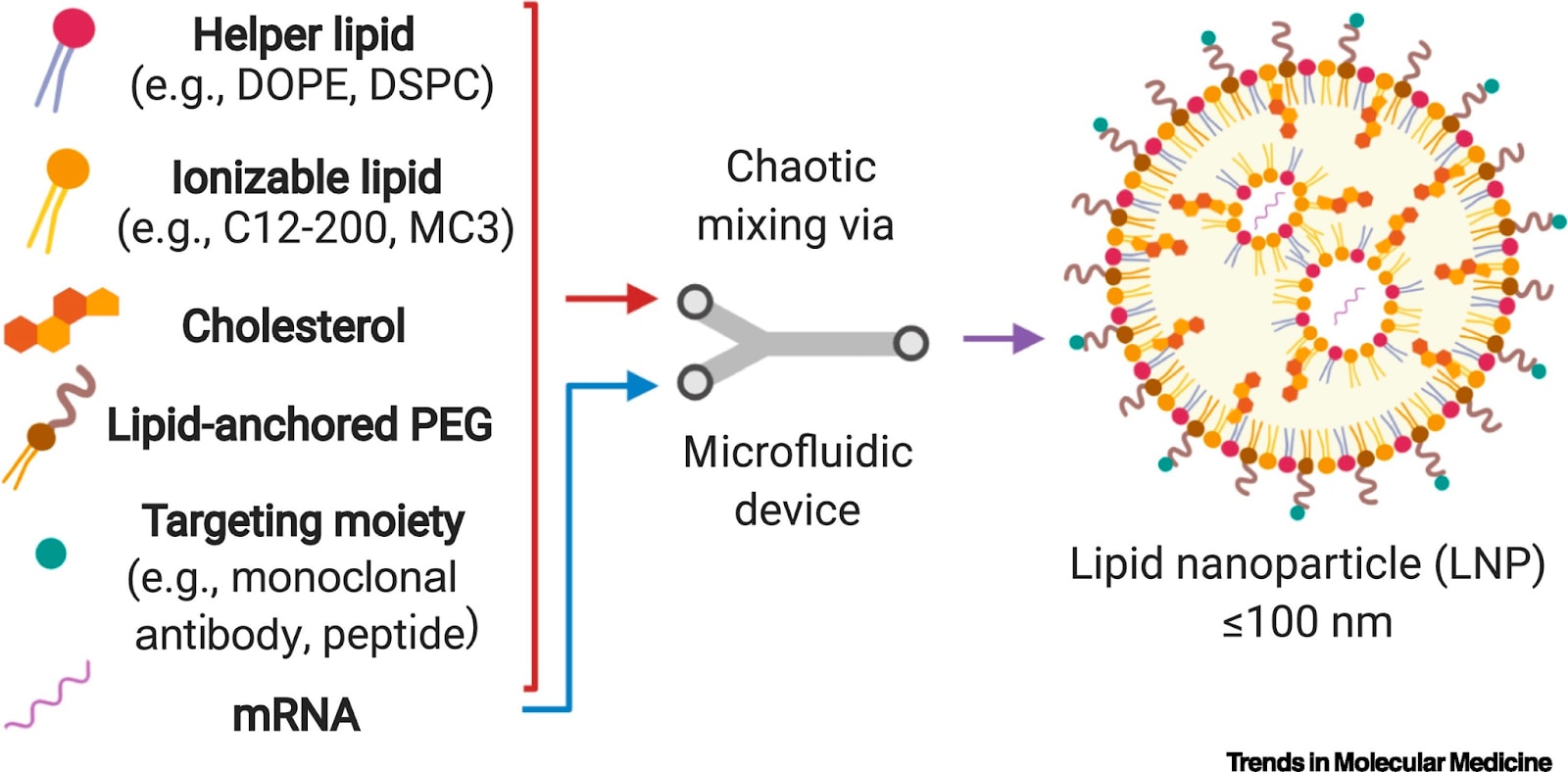
So to get mRNA inside a cell, COVID vaccines stick them inside a delivery container to protect them from the hostile environment outside cells and to get them where they need to go. This container is a little bubble of fat called a lipid nanoparticle.
If you inject these nanoparticles into your muscle tissue, they will float around for a while before being absorbed by your muscle cells via a process called endocytosis.
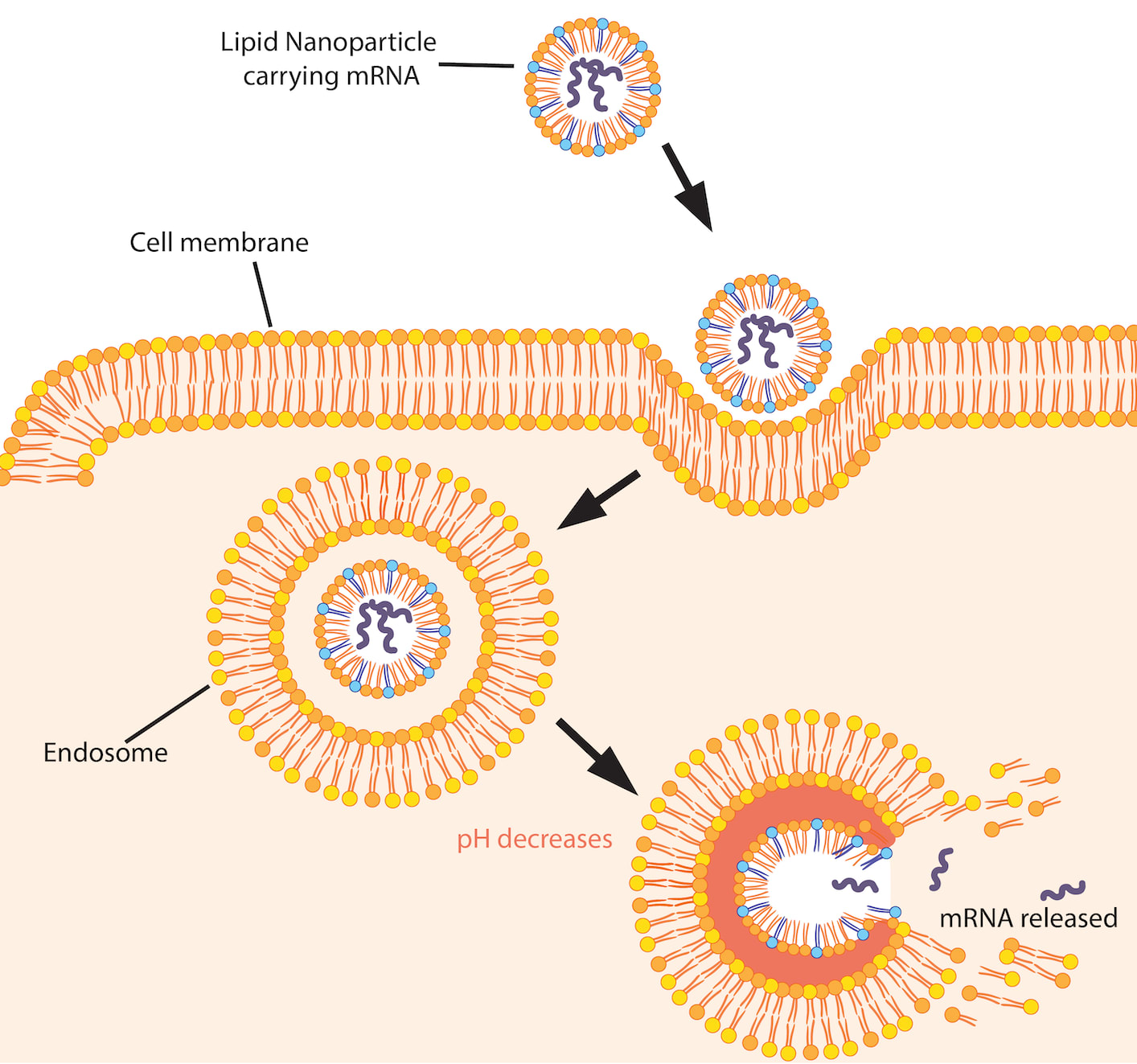
Afterwards, the mRNA is turned into a protein by the ribosomes in the muscle cell. In the case of COVID vaccines the protein is a piece of the virus called the “spike” that SARS-CoV-2 uses to enter your tissues.
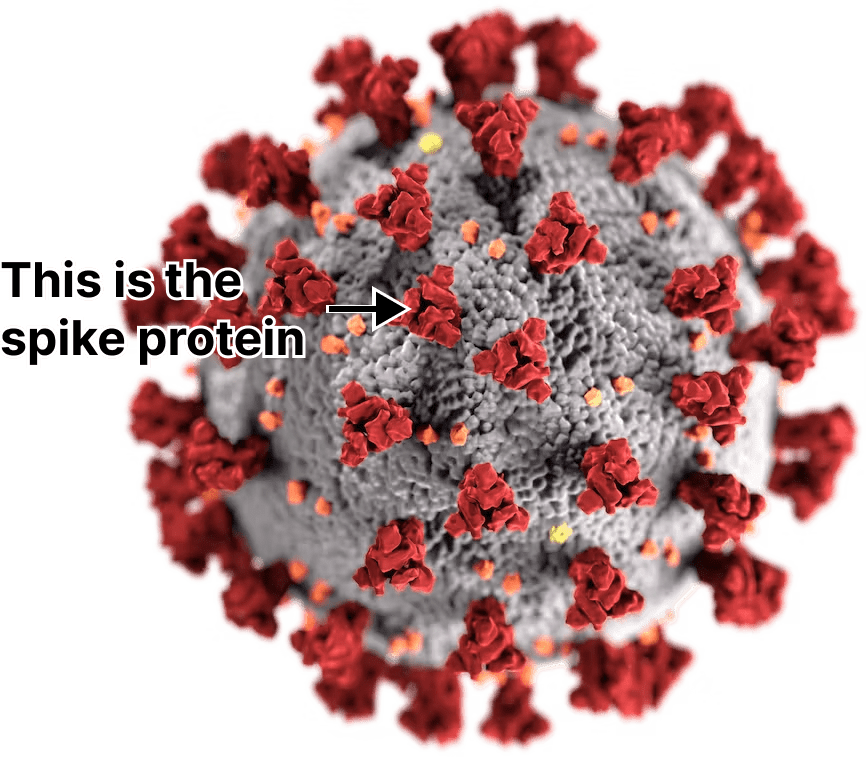
Once created, some of these spike proteins will be chopped up by enzymes and displayed on the outside of each cell’s membrane. Immune cells patrolling the area will eventually bind to these bits of the spike protein and alert the rest of the immune system, triggering inflammation. This is how vaccines teach your body to fight COVID.
So here’s an idea: what if instead of making a spike protein, we deliver mRNA to make a gene editor?
Instead of manufacturing part of a virus, we’d manufacture a CRISPR-based editing tool like a prime editor. We could also ship the guide RNA in the same lipid nanoparticle, which would bond to the newly created editor proteins, allowing them to identify an editing target.
A nuclear localization sequence on the editor would allow the it to enter the cell’s nucleus where the edit could be performed.
So you inject lipid nanoparticles into the brain?
Not exactly. Brains are much more delicate than muscles, so we can’t just copy the administration method from COVID vaccines and inject nanoparticles directly into the brain with a needle.
Well actually we could try. And I plan to look into this during the mouse study phase of the project in case the negative effects are smaller than I imagine.
But apart from issues with possibly damaging brain or spinal tissue (which we could perhaps avoid), the biggest concern with directly injecting editors into the cerebrospinal fluid is distribution. We need to get the editor to all, or at least most cells in the brain or else the effect will be negligible.
If you just inject a needle into a brain, the edits are unlikely to be evenly distributed; most of the cells that get edited will be close to the injection site. So we probably need a better way to distribute lipid nanoparticles evenly throughout the brain.
Thankfully, the bloodstream has already solved this problem for us: it is very good at distributing resources like oxygen and nutrients evenly to all cells in the brain. So the plan is to inject a lipid-nanoparticle containing solution into the patient’s veins, which will flow through the bloodstream and eventually to the brain.

There are two additional roadblocks: getting the nanoparticles past the liver and blood brain barrier.
By default, if you inject lipid nanoparticles into the bloodstream, most of them will get stuck in the liver. This is nice if you want to target a genetic disease in that particular organ (and indeed, there have been studies that did exactly that, treating monogenic high cholesterol in macaques). But if you want to treat anything besides liver conditions then it’s a problem.
From my reading so far, it sounds like nanoparticles circulate through the whole bloodstream many times, and each time some percentage of them get stuck in the liver. So the main strategy scientists have deployed to avoid accumulation is to get them into the target organ or tissue as quickly as possible.
Here’s a paper in which the authors do exactly that for the brain. They attach a peptide called “angiopep-2” onto the outside of the nanoparticles, allowing them to bypass the blood brain barrier using the LRP1 receptor. This receptor is normally used by LDL cholesterol, which is too big to passively diffuse through the endothelial cells lining the brain. But the angiopep-2 molecule acts like a key, allowing the nanoparticles to use the same door.
The paper was specifically examining this for delivery of chemotherapy drugs to glial cells, so it’s possible we’d either need to attach additional targeting ligands to the surface of the nanoparticles to allow them to get inside neurons, or that we’d need to use another ligand besides angiopep. But this seems like a problem that is solvable with more research.
Or maybe you can just YOLO the nanoparticles directly into someone’s brain with a needle. Who knows? This is why research will need to start with cell culture experiments and (if things go well) animal testing.
What if administration via lipid nanoparticles doesn’t work?
If lipid nanoparticles don’t work, there are two alternative delivery vectors we can look into: adeno-associated viruses (AAVs) and engineered virus-like particles (eVLPs).
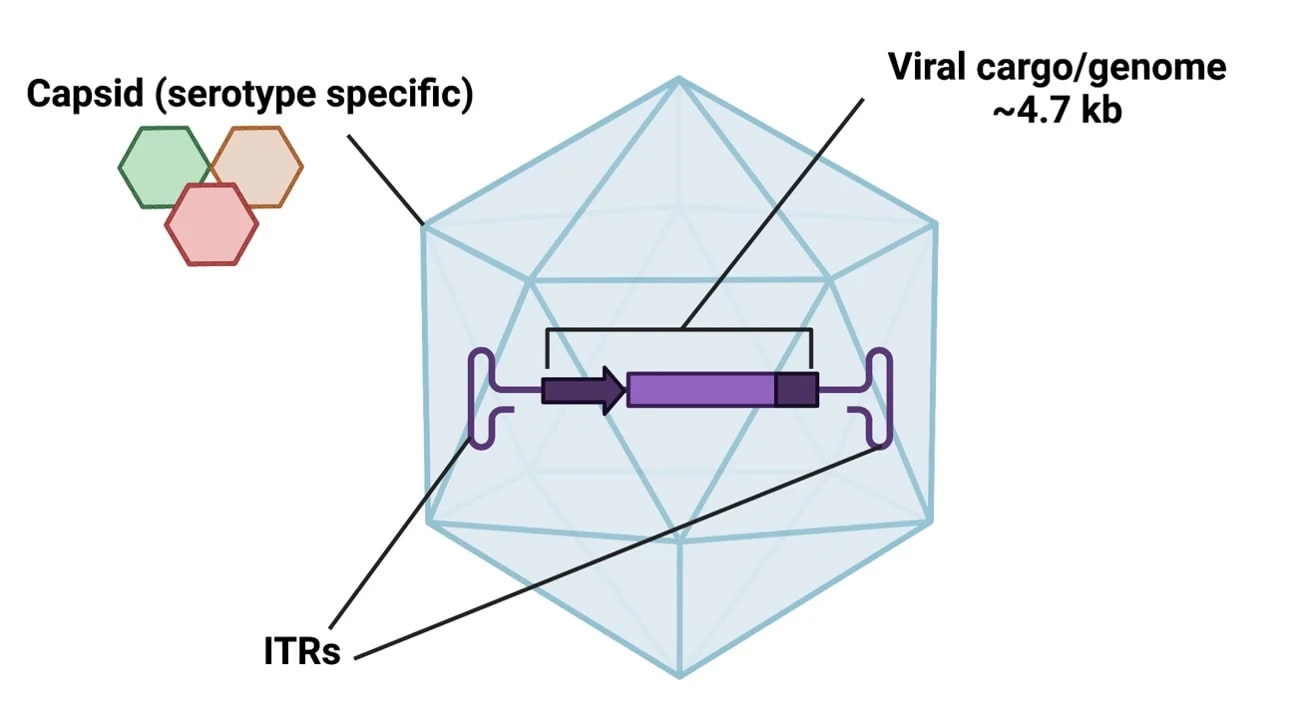
AAVs have become the go-to tool for gene therapy, mostly because viruses are just very good at getting into cells. The reason we don’t plan to use AAVs by default mostly comes down to issues with repeat dosing.
As one might expect, the immune system is not a big fan of viruses. So when you deliver DNA for a gene editor with an AAV, the viral proteins often trigger an adaptive immune response. This means that when you next try to deliver a payload with the same AAV, antibodies created during the first dose will bind to and destroy most of them.
There ARE potential ways around this, such as using a different version of an adenovirus with different epitopes which won’t be detected by antibodies. And there is ongoing work at the moment to optimize the viral proteins to provoke less of an immune response. But by my understanding this is still quite difficult and you may not have many chances to redose.
But there are other viral delivery vectors
Engineered virus-like particles, or eVLPs for short, are another way to get gene editors into cells. Like AAVs, they are constructed of various viral proteins and a payload.
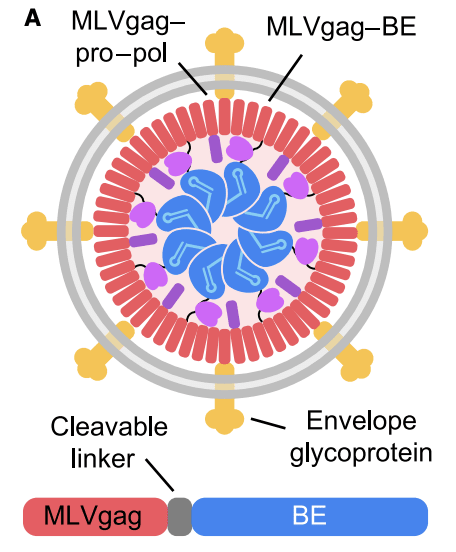
But unlike AAVs, eVLPs usually contain proteins, not DNA. This is important because, once they enter a cell, proteins have a much shorter half-life than DNA. A piece of DNA can last for weeks or sometimes even months inside the nucleus. As long as it is around, enzymes will crank out new editor proteins using that DNA template.
Since most edits are made shortly after the first editors are synthesized, creating new ones for weeks is pointless. At best they will do nothing and at worst they will edit other genes that we don’t want changed. eVLPs fix this problem by delivering the editor proteins directly instead of instructions to make them.
They can also be customized to a surprising degree. The yellow-colored envelope glycoprotein shown in the diagram above can be changed to alter which cells take up the particles. So you can target them to the liver, or the lungs, or the brain, or any number of other tissues within the body.
Like AAVs, eVLPs have issues with immunogenicity. This makes redosing a potential concern, though it’s possible there are ways to ameliorate these risks.
Would edits to the adult brain even do anything?
Apart from the technical issues with making edits, the biggest uncertainty in this proposal is how large of an effect we should expect from modifying genes in adults. All the genetic studies on which our understanding of genes is based assume genes differ FROM BIRTH.
We know that some genes are only active in the womb, or in childhood, which should make us very skeptical that editing them would have an effect. For example, here’s a graph showing the relative level of expression of DCX, a gene active in neuronal progenitor cells and migrating immature neurons.
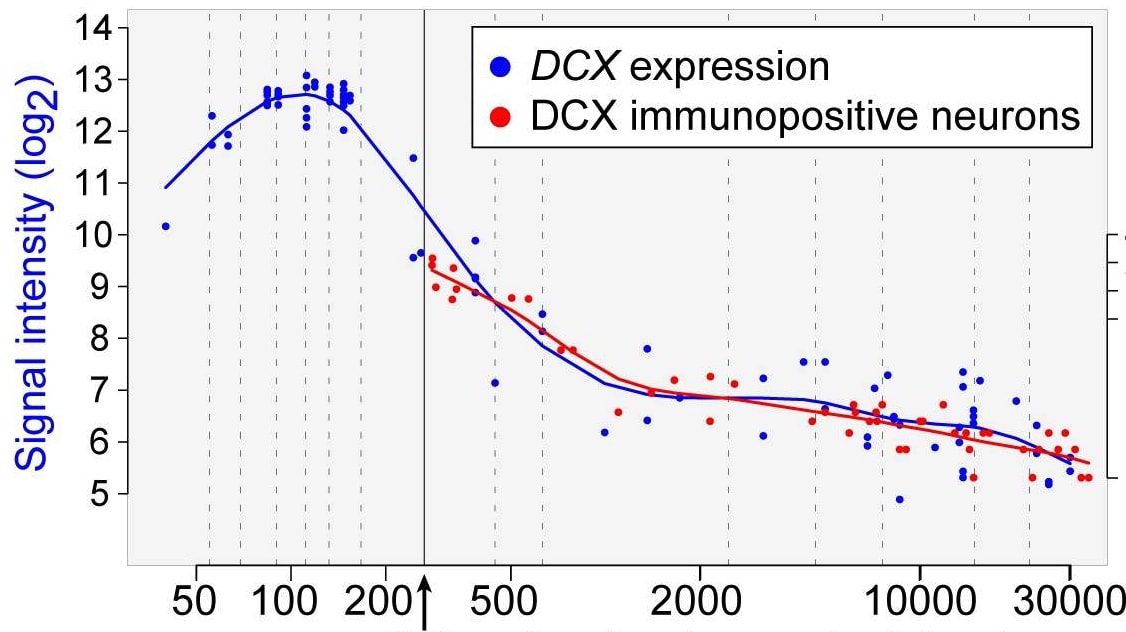
You can see the expression levels peak in the second trimester before dropping by a factor of ~80 by age 27.
We would therefore expect that any changes to promoters or inhibitors of the DCX gene would have little or no effect were they to be made in adulthood.
However, not every gene is like this. Here’s another graph from the same paper showing the relative expression of groups of genes in the hippocampus:
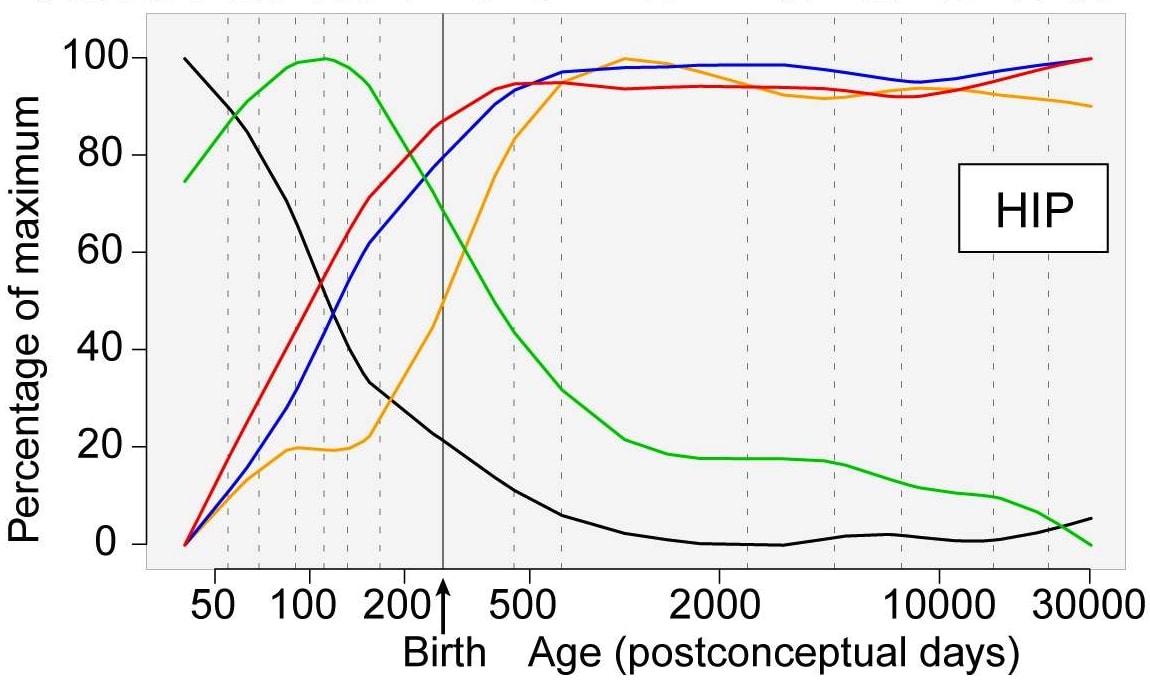
The red, blue and yellow lines, representing the expression of a group of synapse, dendrite, and myelination genes respectively, climb steadily until shortly after birth, then plateau throughout the rest of adulthood. So we could reasonably expect any changes to these genes to have a much stronger effect relative to changes to DEX.
This is one way to develop a basic prior about which genes are likely to have an effect if edited in adults: the size of the effect of a given gene at any given time is likely proportional to its level of expression. Genes that are only active during development probably won’t have as large of an effect when edited.
The paper I linked above collected information about the entire exome, which means we should be able to use its data to form a prior on the odds that editing any given variant will have an effect, though as usual there are some complications with this approach which I address in the appendix.
A systematic analysis of the levels of expressions of all genes throughout the lifespan is beyond the scope of this post, but beyond knowing some proteins reach peak levels in adulthood, here are a few more reasons why I still expect editing to have a large effect:
- We know that about 84% of all genes are expressed somewhere in the adult brain. And while almost none of the intelligence-affecting variants are in protein-coding regions (see the section below), they are almost certainly in regions that AFFECT protein expression, such as promoters and inhibitors. One would naturally expect that modifying such protein expression would have an effect on intelligence.
- Gene therapy in adults ACTUALLY WORKS. We now have several examples of diseases, such as sickle cell anemia, being completely cured in adults through some form of gene therapy.
- We have dozens of examples of traits and diseases whose state is modified by the level of expression of various proteins: Diabetes, Alzheimer’s, autoimmune conditions, skin pigmentation, metabolism, and many others. Since gene editing is ultimately modifying the amount and timing of various proteins, we should expect it to have an effect.
- We know the adult brain can change via forming new connections and changing their strengths. That’s how people learn after all!
What if your editor targets the wrong thing?
Though base editors and prime editors have much lower rates of off-target editing than original CRISPR Cas9, off-target editing is still a concern.
There’s a particularly dangerous kind of off-target edit called a frameshift mutation that we really want to avoid. A frameshift mutation happens when you delete (or insert) a number of base pairs that is not a multiple of 3 from a protein-forming region.
Because base pairs are converted into amino acids three at a time, inserting or deleting a letter changes the meaning of all the base pairs that come after the mutation site.
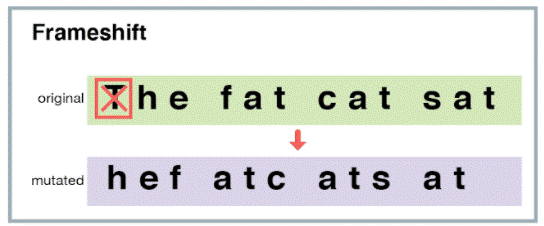
This pretty much always results in a broken protein.
So to minimize the consequences of off-target mutations, we’d do better to target non-protein-coding regions. We already know quite well which regions of the genome are synthesized into proteins. And fortunately, it appears that 98% of intelligence-affecting variants are located in NON-CODING regions. So we should have plenty of variants to target.
What about off-target edits in non-coding regions?
Most of the insertions, deletions and disease-causing variants with small effects are in non-coding regions of DNA. This gives us some evidence that off-target edits made to these regions will not necessarily be catastrophic.
As long as we can avoid catastrophic off-target edits, a few off-targets will be ok so long as they are outweighed by edits with positive effects.
Part of the challenge here is characterizing off-target edits and how they are affected by choice of edit target. If your guide RNA’s binding domain ALMOST matches that of a really critical protein, you may want to think twice before making the edit. Whereas if it’s not particularly close to any target, you probably don’t need to worry as much.
However, it’s still unclear to me how big of a deal off-target edits and indels will be. If you unintentionally insert a few base pairs into the promoter region of some protein, will the promoter just work a little less well, or will it break altogether? I wasn’t able to find many answers in the research literature, so consider this a topic for future research.
It is worth pointing out that long-lived cells in the body such as neurons experience a thousand or more random mutations by age 40 and that, with the exception of cancer, these mutations rarely seem to cause serious dysfunction.
How about mosaicism?
Another potential issue with producing a large number of edits to cells in the brain is cellular mosaicism. The determination of which cell gets which set of edits is somewhat random. Maybe some cells are a little too far away from a blood vessel. Maybe some just don’t get enough of the editor. Regardless of the reason, it’s likely different cells within the brain will receive different sets of edits. Will this cause problems?
It’s possible it will. But as mentioned in the previous section, neurons in the adult brain already have significant genetic differences and most of people seem functional despite these variations. Neurons accumulate about 20-40 single letter mutations per year, with the exact amount varying by brain region and by individual.
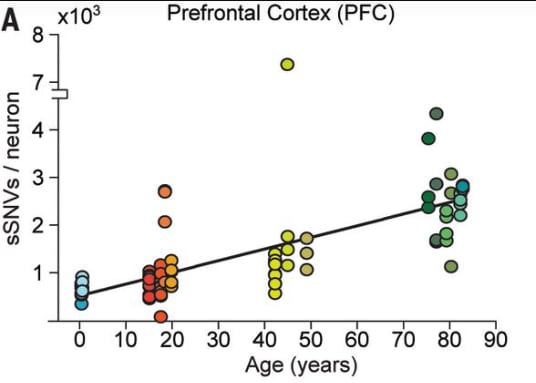
By age 45 or so, the average neuron in an adult brain has about 1500 random genetic mutations. And the exact set of mutations each neuron has is somewhat random.
There are likely to be some differences between mutations caused by gene editing and those caused by cellular metabolism, pollutants, and cosmic rays. In particular, those caused by gene editing are more likely to occur at or near functionally important regions.
Still, it should be at least somewhat comforting that neurons usually remain functional when they have a few thousand genetic differences. But it is possible my optimism is misplaced here and mosaicism will turn out to be a significant challenge for some reason I can’t yet foresee. This is yet another reason to conduct animal testing.
Can we make enough edits in a single cell to have a significant effect?
Making a large enough number of edits to have a significant effect on a phenotype of interest is one of the main challenges with this research protocol. I’ve found fairly recent papers such as Qichen & Xue, that show a reasonably large number of edits can be made in the same cell at the same time; 31 edits using base editors and 3 using prime editors. As with nearly all research papers in this field, they used HEK293T cells, which are unusually easy to transfect with editors. We can therefore treat this as an upper bound on the number of simultaneous edits that can be made with current techniques.
Here’s another research paper from Ni et al. where the researchers used prime editors to make 8 simultaneous edits in regenerated wheat plants.
I also know of a PHD student in George Church’s lab that was able to make several thousand edits in the same cell at the same time by targeting a gene that has several thousand copies spread throughout the genome.
So people are trying multiplex editing, but if anyone has tried to make hundreds of edits at a time to different genes, they don’t seem to have published their results. This is what we need to be able to do for in-vivo multiplex editing to have a big impact on polygenic traits and diseases.
However, we don’t necessarily need to introduce all edits with a single injection. If the delivery vector and the editing process doesn’t provoke an adaptive immune response, or if immunosuppressants can be safely used many times, we can just do multiple rounds of administration; 100 edits during the first round, another 100 in the second round and so on.
If there is an immune response, there are some plausible techniques to avoid adverse reactions during subsequent doses, such as using alternative versions of CRISPR with a different antigenic profile. There are a limited number of these alternative CRISPRs, so this technique is not a general purpose solution.
Making hundreds of edits in cultured cells is the first thing I’d like to focus on. I’ve laid out my plans in more detail in “How do we make this real?”
Why hasn’t someone already done this?
There are hundreds of billions of dollars to be made if you can develop a treatment for brain disorders like Alzheimers, dementia and depression. There’s probably trillions to be made if you can enhance human intelligence.
And yet the people who have developed these tools seem to only be using them to treat rare monogenic disorders. Why?
Part of the answer is that “people are looking into it, but in a pretty limited capacity.” For example, here’s a paper that looked at multiplex editing of immune cells in the context of immunotherapy for cancer. And here’s a post that is strikingly similar to many of the ideas I’ve raised here. I only learned of its existence a week before publishing, but have spoken with the authors who had similar ideas about the challenges in the context of their focus on gene therapy for polygenic disease.
Apart from that I can’t find anything. There are 17 clinical trials involving some kind of CRISPR-based gene editing and I can’t find a single one that targets multiple genes.
So here I’m going to speculate on a couple of reasons why there seems to be so little interest in in-vivo multiplex editing.
Black box biology
To believe multiplex editing can work you have to believe in the power of black box biology. Unlike monogenic diseases, we don’t always understand the mechanism through which genetic variants involved in diabetes or intelligence cause their effects.
You have to believe that it is possible to affect a trait by changing genes whose effect you understand but whose mechanism of action you don’t. And what’s more, you need to believe you can change hundreds of genes in a large number of cells in this manner.
To most people, this approach sounds crazy, unpredictable, and dangerous. But the fact is nature mixes and matches combinations of disease affecting alleles all the time and it works out fine in most cases. There are additional challenges with editing such as off-targets and editing efficiency, but these are potentially surmountable issues.
Vague association with eugenics make some academics shy away
A lot of people in Academia are simply very uncomfortable with the idea that genes can influence outcomes in any context other than extremely simple areas like monogenic diseases. I recently attended a conference on polygenic embryo screening in Boston and the amount of denialism about the role of genetics in things as simple as disease outcomes was truly a sight to behold.
In one particularly memorable speech, Erik Turkheimer, a behavioral geneticist from University of Virginia claimed that, contrary to the findings of virtually every published study, adding more data to the training set for genetic predictors simply made them perform worse. As evidence to support this conclusion, he cited a university study on alcohol abuse with a sample size of 7000 that found no associations of genome-wide significance.
Turkheimer neglected to mention that no one could reasonably expect the study to unearth such associations because it was too underpowered to detect all but the most extreme effects. Most studies in this field have 30-100x the number of participants.
In many cases, arguing against these unsupported viewpoints is almost impossible. Genomic Prediction has published about a dozen papers on polygenic embryo selection, many of which are extremely well written and contain novel findings with significant clinical relevance. Virtually no one pays attention. Even scientists writing about the exact same topics fail to cite their work.
Many of them simply do not care what is true. They view themselves as warriors in a holy battle, and they already know anyone talking about genetic improvement is an evil racist eugenicist.
You can convince an open-minded person in a one-on-one discussion because the benefits are so gigantic and the goal is consistent with widely shared values as expressed in hundreds of other contexts.
But a large portion of Academics will just hear someone say “genes matter and could be improved”, think “isn’t that what Hitler did?”, and become forever unreachable. Even if you explain to them that no, Hitler’s theory of genetics was based on a corrupted version of purity ethics and that in fact a lot of his programs were probably dysgenic, and that even if they weren’t we would never do that because it’s evil, it’s too late. They have already passed behind the veil of ignorance. The fight was lost the moment the association with Hitler entered their mind.
Though these attitudes don’t completely prevent work on gene therapies, I think it likely that the hostility towards altering outcomes through genetic means drives otherwise qualified researchers away from this line of work.
Maybe there’s some kind of technical roadblock I’m missing
I’ve now talked to some pretty well-qualified bio PHDs with expertise in stem cells, gene therapy, and genetics. While many of them were skeptical, none of them could point to any part of the proposed treatment process that definitely won’t work. The two areas they were most skeptical of was the ability to deliver an editing vector to the brain and the ability to perform many edits in the same cell. But both seem addressable. Delivery for one, may be solved by other scientists trying to deliver gene editors to the brain to target monogenic conditions like Huntington’s.
There are unknowns of course, and many challenges which could individually or in conjunction make the project infeasible. For example either of the challenges listed above, or perhaps mosaicism is a bigger deal than I expect it to be.
But I’ve only talked to a couple of people. Maybe someone I haven’t spoken with knows of a hard blocker I am unaware of.
People are busy picking lower hanging fruit
It’s a lot easier to target a specific gene that causes a particular disease than it is to target hundreds or thousands of genes which contribute to disease risk. Given prime editors were only created four years ago and base editors only seven, maybe the people with the relevant expertise are just busy picking lower hanging fruit which has a much better chance of resulting in a money-making therapy. The lowest hanging fruit is curing monogenic diseases, and that’s currently where all the focus is.
The market is erring
Some types of markets are highly efficient. Commodities and stocks, for example, are very efficient because a large number of people have access to the most relevant information and arbitrage can happen extremely quickly.
But the more time I’ve spent in private business, and startups in particular, the more I’ve realized that most industries are not as efficient as the stock market. They are far less open, the knowledge is more concentrated, the entry costs are higher, and there just aren’t that many people with the abilities needed to make the markets more efficient.
In the case of multiplex editing, there are many factors at play which one would reasonably expect to result in a less efficient market:
- The idea of polygenic editing is not intuitive to most people, even some geneticists. One must either hear it or read about it online.
- One must understand polygenic scores and ignore the reams of (mostly) politically motivated articles attempting to show they aren’t real or aren’t predictive of outcomes.
- One must understand gene editing tools and delivery vectors well enough to know what is technically possible.
- One must believe that the known technical roadblocks can be overcome.
- One must be willing to take a gamble that known unknowns and unknown unknowns will not make this impossible (though they may do so)
How many people in the world satisfy all five of those criteria? My guess is not many. So despite how absurd it sounds to say that the pharmaceutical industry is overlooking a potential trillion dollar opportunity, I think there is a real possibility they are. Base editors have been around for seven years and in that time the number of papers published on multiplex editing in human cells for anything besides cancer treatment has remained in the single digits.
I don’t think this is a case of “just wait for the market to do it”.
Can we accelerate development of this technology?
After all that exposition, let me summarize where the work I think will be done by the market and that which I expect them to ignore.
Problems I expect the market to work on
- Targeting editors towards specific organs. There are many monogenic conditions that primarily affect the brain, such as Huntington’s disease or monogenic Alzheimer’s. Companies are already trying to treat monogenic conditions, so I think there’s a decent chance they could solve the issues of efficiently delivering gene editors to brain cells.
Problems the market might solve
- Attenuating the adaptive immune response to editor proteins and delivery vectors.
If you are targeting a monogenic disease, you only need to deliver an editing vector once. The adaptive immune response to the delivery vector and CRISPR proteins is therefore of limited concern; the innate immune response to these is much more important.
There may be overlap between avoiding each of these, but I expect that if an adaptive immune response is triggered by monogenic gene therapy and it doesn’t cause anaphylaxis, companies will not concern themselves with avoiding it. - Avoiding off-target edits in a multiplex editing context.
The odds of making an off-target edit scale exponentially as the number of edits increase. Rare off-targets are therefore a much smaller concern for monogenic gene therapy, and not much of a concern at all for immunotherapy unless they cause cancer or are common enough to kill off a problematic number of cells.
I therefore don’t expect companies or research labs to address this problem at the scale it needs to be addressed within the multiplex editing context.
One possible exception are companies using gene editing in the agricultural space, but the life of a wheat plant or other crop has virtually zero value, so the consequences of off-targets are far less serious.
Problems I don’t expect the market to work on
- Creating better intelligence predictors.
Unless embryo selection for intelligence takes off, I think it is unlikely any of the groups that currently have the data to create strong intelligence predictors (or could easily obtain it) will do so. - Conducting massive multiplex editing of human cells
If you don’t intend to target polygenic disease, there’s little motivation to make multiple genetic changes in cells. And as explained above, I don’t expect many if any people to work on polygenic disease risk because mainstream scientists are very skeptical of embryo selection and of the black box model of biology. - Determining how effective edits are when conducted in adult animals
None of the monogenic diseases targeted by current therapies involve genes that are only active during childhood. It therefore seems fairly unlikely that anyone will do research into this topic since it has no clear clinical relevance.
How do we make this real?
The hardest and most important question in this post is how we make this technology real. It’s one thing to write a LessWrong post pointing out some revolutionary new technology might be possible. It’s quite another to run human trials for a potentially quite risky medical intervention that doesn’t even target a recognized disease.
Fortunately, some basic work is already being done. The first clinical trials of prime editors are likely to start in 2024. And there are at least four clinical trials underway right now testing the efficacy of base editors designed to target monogenic diseases. The VERVE-101 trial is particularly interesting due to its use of mRNA delivered via lipid nanoparticles to target a genetic mutation that causes high cholesterol and cardiovascular disease.
If this trial succeeds, it will definitively demonstrate that in-vivo gene editing with lipid nanoparticles and base editors can work. The remaining obstacles at that point will be editing efficiency and getting the nanoparticles to the brain (a solution to which I’ve proposed in the section titled “So you inject lipid nanoparticles into the brain?”. Here’s an outline of what I think needs to be done, in order:
- Determine if it is possible to perform a large number of edits in cell culture with reasonable editing efficiency and low rates of off-target edits.
- Run trials in mice. Try out different delivery vectors. See if you can get any of them to work on an actual animal.
- Run trials in cows. We have good polygenic scores for cow traits like milk production, so we can test whether or not polygenic modifications in adult animals can change trait expression.
- (Possibly in parallel with cows) run trials on chimpanzees
The goal of such trials would be to test our hypotheses about mosaicism, cancer, and the relative effect of genes in adulthood vs childhood. - Run trials in humans on a polygenic brain disease. See if we can make a treatment for depression, Alzheimer’s, or another serious brain condition.
- If the above passes a basic safety test (i.e. no one in the treatment group is dying or getting seriously ill), begin trials for intelligence enhancement.
It’s possible one or more steps could be skipped, but at the very least you need to run trials in some kind of model organism to refine the protocol and ensure the treatment is safe enough to begin human trials.
My current plan
I don’t have a formal background in biology. And though I learn fairly quickly and have great resources like SciHub and GPT4, I am not going to be capable of running a lab on my own any time soon.
So at the moment I am looking for a cofounder who knows how to run a basic research lab. It is obviously helpful if you have worked specifically with base or prime editors before but what I care most about is finding someone smart that I get along with who will persevere through the inevitable pains of creating a startup. If anyone reading this is interested in joining me and has such a background, send me an email.
I’m hoping to obtain funding for one year worth of research, during which our main goal will be to show we can perform a large number of distinct edits in cell culture. If we are successful in a year the odds of us being able to modify polygenic brain based traits will be dramatically increased.
At that point we would look to either raise capital from investors (if there is sufficient interest), or seek a larger amount of grant funding. I’ve outlined a brief version of additional steps as well as a rough estimate of costs below.
Short-term logistics concerns
A rough outline of my research agenda
- Find a cofounder/research partner
- Design custom pegRNAs for use with prime editors
- Pay a company to synthesize some plasmids containing those pegRNAs with appropriate promoters incorporated
- Rent out lab space. There are many orgs that do this such as BadAss labs, whose facilities I’ve toured.
- Obtain other consumables not supplied by the lab space like HEK cell lines, NGS sequencing materials etc
- Do some very basic validation tests to make sure we can do editing, such as inserting a fluorescent protein into an HEK cell (if the test is successful, the cell will glow)
- Start trying to do prime editing. Assess the relationship between number of edits attempted and editing efficiency.
- Try out a bunch of different things to increase editing efficiency and total edits.
- Try multiple editing rounds. See how many total rounds we can do without killing off cells.
- If the above problems can be solved, seek a larger grant and/or fundraise to test delivery vectors in mice.
Rough estimate of costs
| Cost | Purpose | Yearly Cost | Additional Notes |
| Basic Lab Space | Access to equipment, lab space, bulk discounts | ~66000 | Available from BadAss Labs. Includes workbench, our own personal CO2 incubator, cold storage, a cubicle, access to all the machines we need to do cell culturing, and the facilities to do animal testing at a later date if cell culture tests go well |
| Workers comp & liability coverage | Required by labs we rent space from | ~12,000 | Assuming workers comp is 2% of salaries, 1000 per year for general liability insurance and 3000 per year for professional liability insurance |
| Custom plasmids | gRNA synthesis | 15,000? | I’ve talked to Azenta about the pricing of these. They said they would be able to synthesize the DNA for 7000 custom guide RNAs for about $5000. We probably won’t need that many, but we’ll also likely go through a few iterations with the custom guide RNAs. |
| HEK cell lines & culture media | 2000 | Available from angioproteomie | |
| mRNA for base editors | Creating base editors in cells | 5000? | The cost per custom mRNA is around $300-400 but I assume we'll want to try out many variants |
| mRNA for prime editors | Creating prime editors in cell cultures | 5000? | The cost per custom mRNA is around $300-400 but I assume we'll want to try out many variants |
| Flasks or plates, media, serums, reagents for cell growth, LNP materials etc | Routine lab activity like culturing cells, making lipid nanoparticles and a host of other things | 20,000? | This is a very very rough estimate. I will have a clearer idea of these costs when I find a collaborator with whom I can plan out a precise set of experiments. |
| NGS sequencing | Determining whether the edits we tried to make actually worked | 50,000? | |
| Other things I don't know about | 200,000? | This is a very rough estimate meant to cover lab supplies and services. I will probably be able to form a better estimate when I find a cofounder and open an account with BadAss labs (which will allow me to see their pricing for things) | |
| Travel for academic conferences, networking | I think it will likely be beneficial for one or more of us to attend a few conferences on this topic to network with potential donors. | 20,000? | |
| Salaries | Keeping us off the streets | 400,000 | There will likely be three of us working on this full time |
| Grand Total Estimate | ~795,000 |
Conclusion
Someone needs to work on massive multiplex in-vivo gene editing. This is one of the few game-board-flipping technologies that could have a significant positive impact on alignment prior to the arrival of AGI. If we completely stop AI development following some pre-AGI disaster, genetic engineering by itself could result in a future of incredible prosperity with far less suffering than exists in the world today.
I also think it is very unlikely that this will just “happen anyways” without any of us making an active effort. Base editors have been around since 2016. In that time, anyone could have made a serious proposal and started work on multiplex editing. Instead we have like two academic papers and one blog post.
In the grand scheme of things, it would not take that much money to start preliminary research. We could probably fund myself and my coauthor, a lab director, and lab supplies to do research for a year for under a million dollars. If you’re interested in funding this work and want to see a more detailed breakdown of costs than what I’ve provided above, please send me an email.
I don’t know exactly how long it will take to get a working therapy, but I’d give us a 10% chance of having something ready for human testing within five years. Maybe sooner if we get more money and things go well.
Do I think this is more important than alignment work?
No.
I continue to think the most likely way to solve alignment is for a bunch of smart people to work directly on the problem. And who knows, maybe Quintin Pope and Nora Belrose are correct and we live in an alignment by default universe where the main issue is making sure power doesn’t become too centralized in a post-AGI world.
But I think it’s going to be a lot more likely we make it to that positive future if we have smarter, more focused people around. If you can help me make this happen through your expertise or funding, please send me a DM or email me.
Appendix (Warning: boredom ahead)
Additional headaches not discussed
Regarding editing
Bystander edits
If you have a sequence like “AAAAA” and you’re trying to change it to “AAGAA”, a base editor will pretty much always edit it to “GGGGG”. This is called “bystander editing” and will be a source of future headaches. It happens because the deaminase enzyme is not precise enough to only target exactly the base pair you are trying to target; there’s a window of bases it will target. When I’ve written some software to identify which genetic variants we want to target I will be able to give a better estimate of how many edits we can target with base editors without inducing bystander edits. There are papers that have reduced the frequency of bystander edits, such as Gehrke et al, but it’s unclear to me at the moment whether the improvement is sufficient to make base editors usable on such sequences.
Chemical differences of base editor products
Traditional base editors change cytosine to uracil and adenine to inosine. Many enzymes and processes within the cell treat uracil the same as thymine and inosine the same as guanine. This is good! It’s why base editors work in the first place. But the substituted base pairs are not chemically identical.
- They are less chemically stable over time.
- There may be some differences in how they are treated by enzymes that affect protein synthesis or binding affinity.
It’s not clear to me at this moment what the implications of this substitution are. Given there are four clinical trials underway right now which use base editors as gene therapies, I don’t think they’re a showstopper; at most, I think they would simply reduce editing efficiency. But these chemical differences are something we will need to explore more during the research process.
Editing silenced DNA
There is mixed information online about the ability of base and prime editors to change DNA that is methylated or wrapped around histones. Some articles show that you can do this with modified base editors, but imply normal base editors have limited ability to edit such DNA. Also the ability seems to depend on the type of chromatin and the specific editor.
Avoiding off-target edits
Base editors (and to a lesser extent prime editors) have issues with off-target edits. The rate of off-targets depends on a variety of factors, but one of the most important is the similarity between the sequence of the binding site and that of other sites in the genome. If the binding site is 20 bases long, but there are several other sequences in the genome that only differ by one letter, there’s a decent chance the editor will bind to one of those other sequences instead.
But quantifying the relationship between the binding sequence and the off-target edit rate is something we still need to do for this project. We can start with models such as Deepoff, DeepCRISPR, Azimuth/Elevation, and DeepPrime to select editing targets (credit to this post for bringing these to my attention), then validate the results and possibly make adjustments after performing experiments.
Regarding fine-mapping of causal variants
There’s an annoying issue with current data on intelligence-affecting variants that reduces the odds of us editing the correct variant. As of this writing, virtually all of the studies examining the genetic roots of intelligence used SNP arrays as their data source. SNP arrays are a way of simplifying the genome sequencing process by focusing on genes that commonly vary between people. For example, the SNP array used by UK Biobank measures all genetic regions where at least ~1% of the population differs.
This means the variants that are implicated as causing an observed intelligence difference will always come from that set, even if the true causal variant is not included in the SNP array. This is annoying, because it sometimes means we’ll misidentify which gene is causing an observed difference in intelligence.
Fortunately there’s a relatively straightforward solution: in the short run we can use some of the recently released whole genome sequences (such as the 500k that were recently published by UK biobank) to adjust our estimates of any given variant causing the observed change. In the long run, the best solution is to just retrain predictors using whole genome data.
Regarding differential gene expression throughout the lifespan
Thanks to Kang et al. we have data on the level of expression of all proteins in the brain throughout the lifespan. But for reasons stated in “What if your editor targets the wrong thing”, we’re planning to make pretty much all edits to non-protein coding regions. We haven’t yet developed a mechanistic plan for linking non-coding regions with the proteins whose expression they affect. There are some cases where this should be easy, such as tying a promoter (which is always directly upstream of the exonic region) to a protein. But in other cases such as repressor genes which aren’t necessarily close to the affected gene, this may be more difficult.
Regarding immune response
I am not currently aware of any way to ensure that ONLY brain cells receive lipid nanoparticles. It’s likely they will get taken up by other cells in the body, and some of those cells will express editing proteins and receive edits. These edits probably won’t do much if we are targeting genes involved in brain-based traits, but non-brain cells do this thing called “antigen presentation” where they chop up proteins inside of themselves and present them on their surfaces.
It’s plausible this could trigger an adaptive immune response leading to the body creating antibodies to CRIPSR proteins. This wouldn’t necessarily be bad for the brain, but it could mean that subsequent dosings would lead to inflammation in non-brain regions. We could potentially treat this with an immunosupressant, but then the main advantage of lipid nanoparticles being immunologically inert kind of goes away.
Regarding brain size
Some of the genes that increase intelligence do so by enlarging the brain. Brain growth stops in adulthood, so it seems likely that any changes to genes involved in determining brain size would simply have no effect in adults. But it has been surprisingly hard to find a definitive answer to this question.
If editing genes involved in brain growth actually restarted brain growth in adulthood AFTER the fusion of growth plates in the cranium, it would cause problems.
If such a problem did occur we could potentially address it by Identifying a set of genes associated with intelligence and not brain size and exclusively edit those. We have GWAS for brain size such as this one, though we would need to significantly increase the data set size to do this.
But this is a topic we plan to study in more depth as part of our research agenda because it is a potential concern.
Editing induced amnesia?
Gene editing in the brain will change the levels and timings of various proteins. This in turn will change the way the brain learns and processes information. This is highly speculative, but maybe this somehow disrupts old memories in a way similar to amnesia.
I’m not very concerned about this because the brain is an incredibly adaptable tissue and we have hundreds of examples of people recovering functionality after literally losing large parts of their brain due to stroke or trauma. Any therapeutic editing process that doesn’t induce cancer or break critical proteins is going to be far, far less disruptive than traumatic brain injuries. But it’s possible there might be a postoperative recovery period as people’s brains adjust to their newfound abilities.
An easy way to de-risk this (in addition to animal trials) is by just doing a small number of edits during preliminary trials of intelligence enhancement.
Model assumptions
The model for gain as a function of number of edits was created by Kman with a little bit of feedback from GeneSmith.
It makes a few assumptions:
- We assume there are 20,000 IQ-affecting variants with an allele frequency of >1%. This seemed like a reasonable estimation to me based on a conversation I had with an expert in the field, though there are papers that put the actual number anywhere between 10,000 and 24,000.
- We use an allele frequency distribution given by the “neutral and nearly neutral mutation” model, as explained in this paper. There is evidence of current negative selection for intelligence in developed countries and evidence of past positive selection for higher-class traits (of which intelligence is almost certainly one), the model is fairly robust to effects of this magnitude.
- We assume 10 million variants with minor allele frequency >1% (this comes directly from UK Biobank summary statistics, so we aren’t really assuming much here)
- We assume SNP heritability of intelligence of 0.25. I think it is quite likely SNP heritability is somewhat higher, so if anything our model is conservative along this dimension
- The effect size distribution is directly created using the assumption above. We assume a normal distribution of effect sizes with a mean of zero and variance set to match the estimated SNP heritability. Ideally our effect size distribution would more closely match empirical data, but this sort of effect size distribution is commonly used by other models in the field, and since it was easier to work with we used it too. But consider this an area for future improvement.
- We use a simplified LD model with 100 closely correlated genetic variants. The main variable that matters here is the correlation between the MOST closely correlated SNP and the one causing the effect. We eyeballed some LD data to create a uniform distribution from which to draw the most tightly linked variant. So for example, the most tightly correlated SNP might be a random variable whose correlation is somewhere between .95 and 1. All other correlated variants are decrements of .01 less than that most correlated variant. So if the most tightly correlated variant has a correlation of .975, then the next would be correlated at .965, then .955, .945 etc for 100 variants.
This is obviously not a very realistic model, but it probably produces fairly realistic results. But again, this is an area for future improvement.
Editor properties
Here’s a table Kman and I have compiled showing a good fraction of all the base and prime editors we’ve discovered with various attributes.
| Editor | Edits that can be performed | On-target efficiency in HEK293T cells | Off-target activity in HEK293T cells | On-target edit to indel ratio | Activity window width |
| ABE7.10 | A:T to G:C | ~50% | <1% | >500 | ~3 bp |
| ABEmax | ~80% | ? | ~50 | ? | |
| BE2 | G:C to A:T | ~10% | ? | >1000 | ~5 bp |
| BE3 | ~30% | ? | ~23 | ||
| BE4max | ~60% | ? | ? | ||
| TadCBE | C:G to TA | ~57% | 0.38-1.1% | >83 | ? |
| CGBE1 | G:C to C:G | ~15% | ? | ~5 | ~3 bp |
| ATBE | A:T to T:A | ? | ? | ? | ? |
| ACBE-Q | A:T to C:G | ? | ? | ? | ? |
| PE3 | Short (~50 bp) insertions, deletions, and substitutions | ~24% | <1% | ~4.5 | N/A
|
| PE3b | 41% | ~52 | |||
| PE4max | ~20% | ~80 | |||
| PE5max | ~36% | ~16 | |||
| Cas9 + HDR | Insertions, deletions, substitutions | ~0.5% | ~14% | 0.17 |
- Note that these aren’t exactly apples to apples comparisons since editing outcomes vary a lot by edit site, and different sites were tested in different studies (necessarily, since not all editor variants can target the same sites)
- Especially hard to report off-target rates since they’re so sequence dependent
- On-target editing is also surprisingly site-dependent
- Something that didn’t make the table: unintended on-target conversions for base editors
- E.g. CBE converting G to C instead of G to A
- Some of the papers didn’t report averages, so I eyeballed averages from the figures
If you’ve made it this far send me a DM. I want to know what motivates people to go through all of this.

{kind=link}
Yes. Once this tech works you can use it for basically anything so long as you can make enough edits and the genes involved are known.
So we really don’t need to make any special considerations for memory.